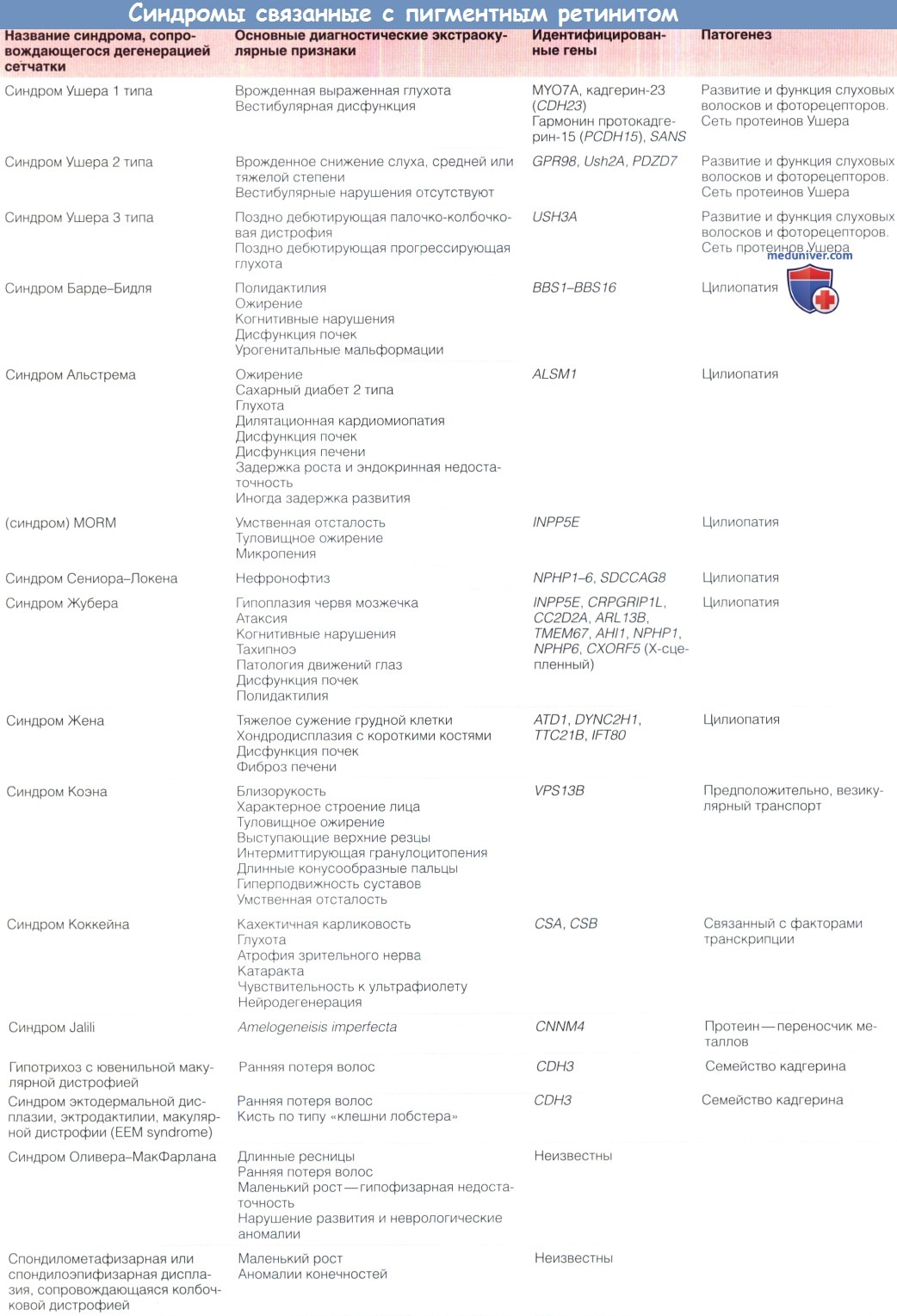
Синдром ушера и 2 группа

Синдром Ушера (иногда синдром Ашера, англ. Usher syndrome, врожденная нейросенсорная глухота и пигментный ретинит) — сравнительно редкое генетическое заболевание, вызываемое мутацией одного из 10 генов, приводящее к врождённой нейросенсорной тугоухости и прогрессирующей потере зрения (пигментная дегенерация). Одна из основных причин слепоглухоты. В настоящее время неизлечим. Наследуется по аутосомно-рецессивному принципу.
Синдром Ушера — наиболее распространённая болезнь, приводящая к слепоглухоте[1][2]. В США с синдромом Ушера рождаются четверо из 100 тысяч новорожденных[3]. В Великобритании, по состоянию на 2010 год, проживали 132 тысячи слепоглухих — примерно 0,2 % населения[4].
Фоторецепторные клетки обычно дегенерируют от периферии к центру сетчатки, включая макулу (жёлтое пятно). Эта дегенерация сначала проявляется как куриная слепота (ослабление зрения при слабом освещении). Периферическое зрение постепенно теряется, ограничивая поле зрения до туннельного, постепенно приводя к полной слепоте[5].
Существует несколько[6][7][8][9][10] классификаций синдрома Ушера, но наиболее распространённой является классификация по степени глухоты[11].
- I тип, встречается с частотой 3-6 на 100 тысяч человек, сопровождается врождённой глубокой тугоухостью или полной глухотой и нарушением вестибулярных функций, раннее начало пигментного ретинита. Связан с мутациями в генах CDH23, MYO7A, PCDH15, USH1C, USH1G. Более распространён среди евреев-ашкеназов.
- II тип, сопровождается тугоухостью, не ухудшающейся со временем, вестибулярные функции не нарушены. Пигментная дегенерация сетчатки начинается с возраста 10-20 лет. Связан с мутациями генов USH2A, GPR98, DFNB31.
- III тип, прогрессирующее ухудшение слуха и зрения, примерно в половине случаев сопровождается вестибулярными нарушениями. Чаще встречается у финнов. Связан с мутациями гена CLRN1.
Литература[править | править код]
- C. H. Usher. On the inheritance of retinitis pigmentosa with notes of cases. Royal London Ophthalmological Hospital Report, 1914; 19: 130—336.
- Stiefel S. H., Lewis R. A. The Madness of Usher’s: Coping With Vision and Hearing Loss/Usher Syndrome Type II (англ.). — Business of Living Publications, 1991. — ISBN 978-1-879518-06-3.
- Duncan E., Prickett H. T. Usher’s Syndrome: What It Is, How to Cope, and How to Help (англ.). — Charles C. Thomas, 1988. — ISBN 978-0-398-05481-6.
- Vernon M. Answers to your questions about Usher’s syndrome (retinitis pigmentosa with hearing loss) (англ.). — Foundation Fighting Blindness, 1986.
- Vernon M. Usher’s syndrome: Deafness and progressive blindness : clinical cases, prevention, theory and literature survey (англ.). — Pergamon Press (англ.)русск., 1969.
Примечания[править | править код]
- ↑ Vernon M. Usher’s syndrome—deafness and progressive blindness. Clinical cases, prevention, theory and literature survey. (англ.) // Journal of chronic diseases. — 1969. — Vol. 22, no. 3. — P. 133—151. — doi:10.1016/0021-9681(69)90055-1. — PMID 4897966.
- ↑ Балашова Л.М., Гонтаренко Ю.Э. и др. Положение людей с выраженными нарушениями слуха и зрения (слепоглухих) в Российской Федерации. Отчёт по результатам исследования (pdf) (недоступная ссылка). Институт политики детства и прикладной социальной работы (2015). Дата обращения 1 апреля 2017. Архивировано 13 июля 2017 года.
- ↑ American deaf-blind population (англ.). LibGuides (July 2010). Дата обращения 1 апреля 2017.
- ↑ Estimating the Number of People with Co‐Occurring Vision and Hearing Impairments in the UK (англ.) (pdf) (недоступная ссылка). Centre for Disability Research (April 2010). Дата обращения 1 апреля 2017. Архивировано 1 апреля 2017 года.
- ↑ Williams D. S. Usher syndrome: animal models, retinal function of Usher proteins, and prospects for gene therapy. (англ.) // Vision research. — 2008. — Vol. 48, no. 3. — P. 433—441. — doi:10.1016/j.visres.2007.08.015. — PMID 17936325.
- ↑ Gorlin R. J., Tilsner T. J., Feinstein S., Duvall A. J. 3rd. Usher’s syndrome type III. (англ.) // Archives of otolaryngology (Chicago, Ill. : 1960). — 1979. — Vol. 105, no. 6. — P. 353—354. — doi:10.1001/archotol.1979.00790180051011. — PMID 454290.
- ↑ Merin S., Auerbach E. Retinitis pigmentosa. (англ.) // Survey of ophthalmology. — 1976. — Vol. 20, no. 5. — P. 303—346. — doi:10.1016/S0039-6257(96)90001-6. — PMID 817406.
- ↑ Fishman G. A., Kumar A., Joseph M. E., Torok N., Anderson R. J. Usher’s syndrome. Ophthalmic and neuro-otologic findings suggesting genetic heterogeneity. (англ.) // Archives of ophthalmology (Chicago, Ill. : 1960). — 1983. — Vol. 101, no. 9. — P. 1367—1374. — doi:10.1001/archopht.1983.01040020369005. — PMID 6604514.
- ↑ Sankila E. M., Pakarinen L., Kääriäinen H., Aittomäki K., Karjalainen S., Sistonen P., de la Chapelle A. Assignment of an Usher syndrome type III (USH3) gene to chromosome 3q. (англ.) // Human molecular genetics. — 1995. — Vol. 4, no. 1. — P. 93—98. — doi:10.1093/hmg/4.1.93. — PMID 7711740.
- ↑ HALLGREN B. Retinitis pigmentosa combined with congenital deafness; with vestibulo-cerebellar ataxia and mental abnormality in a proportion of cases: A clinical and genetico-statistical study. (англ.) // Acta psychiatrica Scandinavica. Supplementum. — 1959. — Vol. 34, no. 138. — P. 1—101. — doi:10.1111/j.1600-0447.1959.tb08605.x. — PMID 14399116.
- ↑ Smith R. J., Berlin C. I., Hejtmancik J. F., Keats B. J., Kimberling W. J., Lewis R. A., Möller C. G., Pelias M. Z., Tranebjaerg L. Clinical diagnosis of the Usher syndromes. Usher Syndrome Consortium. (англ.) // American journal of medical genetics. — 1994. — Vol. 50, no. 1. — P. 32—38. — doi:10.1002/ajmg.1320500107. — PMID 8160750.
Ссылки[править | править код]
- Ирина Кравцова. Не лечится, помочь нельзя.. Что такое синдром Ушера и как живут слепоглухие в России.. Meduza (31 марта 2017). Дата обращения 1 апреля 2017.
- Что такое синдром Ушера? // по материалам Конференции по проблемам слепоглухоты и синдрома Ушера, март 1999, Москва.
- СИНДРОМ УШЕРА // «ОБЪЕДИНЕНИЕ СЛАБОСЛЫШАЩИХ»
Источник
Синдром Ушера (Usher’s syndrome, USH) — глухой слепнущий ребенокСиндром Ушера (Usher’s syndrome, USH) — комбинация сенсоневральной глухоты с прогрессирующей дегенерацией сетчатки. Синдромом Ушера страдает один из каждых десяти глухих детей (5% всех случаев врожденной глухоты). Это наиболее часто встречающийся наследственный синдром, сопровождающийся глухотой и наиболее часто встречающийся синдром среди слепоглухонемых. Синдром подразделяется на три группы: синдром Ушера 1 типа (USH1), 2 типа (USH2) и 3 типа (USH3). Синдром Ушера 1 и 2 типов обычно диагностируется в раннем или старшем детском возрасте, соответственно, но может диагностироваться и позже, поскольку ухудшение зрения у глухого ребенка может оставаться незамеченным. Вестибулярная дисфункция связана с классической картиной синдрома Ушера 1 типа. Каждый тип синдрома Ушера наследуется по аутосомно-рецессивному механизму, но наблюдается генетическая гетерогенность этих заболеваний. Для каждого типа идентифицировано несколько генов. С помощью молекулярных исследований выявлено клиническое частичное совпадение классических первого и второго типов синдрома Ушера. Некоторые гены Ушера могут вызывать развитие изолированных глухоты или пигментного ретинита (USH2A). Таким детям настоятельно рекомендуется применение кохлеарных имплантов (особенно при USH1). Лечение дегенерации сетчатки все еще не разработано. а) Синдром Ушера 1 типа: наиболее тяжелая форма. Синдром Ушера 1 типа проявляется у детей тяжелой или полной сенсоневральной глухотой, ребенок поздно начинает садиться и ходить вследствие нарушения вестибулярной функции. В раннем детстве развивается дегенерация сетчатки. Уже в возрасте 2-3 лет изменения ЭРГ подтверждают диагноз дегенерации сетчатки, хотя глазное дно выглядит нормальным. Известно пять генов, мутации которых вызывают развитие заболевания, их частота варьирует в зависимости от исследуемой популяции: MY07A (считается основным геном, вызывающим синдром Ушера 1 типа, его мутации выявляются в половине случаев; также мутации этого гена вызывают несиндромальную глухоту), USH1C, CDH23, PCDH15 и USH1G. Хотя заболевание всегда протекает тяжело, его течение может различаться в зависимости от типа мутации. б) Синдром Ушера 2 типа: более позднее развитие дегенерации сетчатки у глухого ребенка. Врожденная сенсоневральная стабильная глухота от легкой до тяжелой степени тяжести, выраженные нарушения в области высоких частот и нормальная вестибулярная функция — основные признаки синдрома Ушера 2 типа. Дегенерация сетчатки развивается позже, чем при синдроме Ушера 1 типа, обычно в пубертатном периоде или позже. Синдром Ушера 2 типа и синдром Ушера 3 типа обычно не проявляются в течение первого десятилетия жизни и первоначально не вызывают развития изменений глазного дна. У многих пациентов сохраняется хорошая острота зрения, несмотря на сужение полей зрения. На ЭРГ на ранних стадиях изменения могут отсутствовать; однако изменения ЭРГ могут выявляться у детей и до развития клинической картины. Во втором десятилетии жизни болезнь проявляется ночной слепотой и утратой периферического зрения и необратимо прогрессирует. Идентифицировано три гена: USH2A (при изолированном пигментном ретините также выявлен мутантный ушерин), GPR98 и DFNB31. в) Синдром Ушера 3 типа: более поздний дебют, течение вариабельно. Возраст дебюта пигментного ретинита при синдроме Ушера 3 типа вариабелен, изменения сетчатки часто выявляются в возрасте старше двадцати лет, но могут быть ошибочно приняты за проявления двух других типов заболевания. Сенсорная глухота развивается уже постлингвально (в отличие от прелингвального развития глухоты при синдроме Ушера 1 и 2 типов), речь у таких пациентов развита нормально, но болезнь прогрессирует и приводит к полной слепоте. Вестибулярная дисфункция различной тяжести развивается у половины пациентов. Ген синдрома Ушера 3 типа особенно часто выявлялся у евреев-ашкенази и финнов. У пациентов с синдромом Ушера 1 и 2 типа также обнаружен мутантный USH3. г) Патогенез синдрома Ушера. В изучении патогенеза этого заболевания достигнут прогресс, установлено, что поражаются слуховые волоски внутреннего уха и фоторецепторные клетки сетчатки. Продукты гена USH1 формируют сеть, интерактом Ушера, который играет ключевую роль в раннем развитии пучков слуховых волосков. Они также являются ключевым компонентом механизма механоэлектрической трансдукции, необходимого для реализации функции слуха. В сетчатке интерактом Ушера действует на уровне цилиарной/пери-цилиарной зоны, в области соединения соединительного волоска с фоторецепторной клеткой. Эти структуры играю важную роль в транспорте протеинов между наружными и внутренними сегментами. Дефекты протеинов USH вызывают рано дебютирующую дисфункцию как внутреннего уха, так и сетчатки.
— Вернуться в содержание раздела «офтальмология» на сайте Оглавление темы «Наследственная патология сетчатки.»:
|
Источник
 Синдром Ашера (Ушера, англ. Usher syndrome, врожденная нейросенсорная глухота и пигментный ретинит) представляет собой аутосомно-рецессивное заболевание, включающее в себя зрительные (пигментный ретинит) и слуховые/вестибулярные нарушения. Распространенность синдрома Ашера варьирует от 4 до 17 на 100 000 человек, представлен тремя клиническими подтипами: USH1, USH2 и USH3, которые различаются степенью тяжести нарушений слуха, наличием или отсутствием вестибулярной дисфункции и началом развития пигментного ретинита.
Синдром Ашера (Ушера, англ. Usher syndrome, врожденная нейросенсорная глухота и пигментный ретинит) представляет собой аутосомно-рецессивное заболевание, включающее в себя зрительные (пигментный ретинит) и слуховые/вестибулярные нарушения. Распространенность синдрома Ашера варьирует от 4 до 17 на 100 000 человек, представлен тремя клиническими подтипами: USH1, USH2 и USH3, которые различаются степенью тяжести нарушений слуха, наличием или отсутствием вестибулярной дисфункции и началом развития пигментного ретинита.
Еще в 1858 г. Von Graefe сообщил о 5 случаях пигментного ретинита с тугоухостью с предполагаемой рецессивной формой наследования. Синдром Ушера — редко наблюдаемое заболевание; в общей популяции он встречается с частотой 3 случая на 100 000. Этот синдром диагностируют у 5-8 % больных с наследственной глухотой. Поскольку частота носительства мутантного гена составляет 1 на 100 человек при рецессивном типе наследования, этот синдром манифестирует редко. Точность диагностики возросла с появлением квантитативной аудиометрии. Истинный синдром Ушера характеризуется грубой наследственной сенсорно-нейрональной стабильной (возможно, прогрессирующей) потерей слуха в сочетании с пигментной ретинопатией, не отличимой от пигментного ретинита, которая сопровождается значительным затруднением речи.
Характерными симптомами синдрома Ушера являются ночная слепота и сужение полей зрения, возникающие в возрасте от 5 до 15 лет. Некоторые авторы считают синдром Ушера генетически гетерогенным, другие — фено-типическим проявлением пигментного ретинита. Предполагают, что среди патогенетических механизмов этого заболевания, передаваемого по аутосомно-рецессивному типу, имеется множество трофических нарушений, которые не могут быть определены одним геном. Однако возникновение глазных симптомов связывают с мутацией гена родопсина. Анализ сцепления указывает на локализацию дефекта в длинном плече хромосомы 4 в области 4(q11- q13).
 В настоящее время синдром Ушера — группа генетически гетерогенных заболеваний, наследуемых аутосомно-рецессивно. По клиническим проявлениям выделяют три основных типа заболевания, причем первые два типа включают несколько форм.
В настоящее время синдром Ушера — группа генетически гетерогенных заболеваний, наследуемых аутосомно-рецессивно. По клиническим проявлениям выделяют три основных типа заболевания, причем первые два типа включают несколько форм.
- Первый тип синдрома Ушера представлен 6 формами.
- При анализе сцепления было показано, что первая форма, названная USH1A, локализуется в районе длинного плеча хромосомы 14 (q32.1- q32.3) между локусами D14S78 и D14S250, но ген пока не клонирован.
- Вторая форма, названная USH1B, является наиболее распространенной: ее диагностируют примерно у 75 % от общего количества больных с первым типом заболевания. Эту форму удалось локализовать на длинном плече хромосомы 11q13.5, в районе ДНК-маркера DUS533. В этом же районе был клонирован один из генов миозина типа VILA. Скрининг этого гена на возможные мутации показал их наличие, основная масса которых приводила к преждевременному терминированию синтеза белка.
- Третья форма первого типа — USH1C была картирована в районе короткого плеча хромосомы 11р15.1 и связана с маркером D11S419. В гене-кандидате, названном гармонином, были обнаружены мутации, приводящие к синтезу укороченного белка, в котором отсутствовали его основные домены.
- USH1D удалось картировать в районе длинного плеча хромосомы 10 между локусами D10S529 и D10S573.
- USH1E — сцепление с ДНК-маркерами D21S1905 и D21S1913, расположенными в длинном плече хромосомы 21q21. Генетическое расстояние между этими маркерами составляет около 15 сМ (сантиморган).
- USH1F, с помощью геномного скрининга была картирована на хромосоме 10 в районе локусов D10S199 и D10S596.
- Второй тип синдрома Ушера включает 2 формы.
- Первая из них — USH2A была картирована в районе длинного плеча хромосомы 1q41 между ДНК-маркерами D1S237 и D1S229. Был клонирован ген-кандидат, названный ушерином. Ген USH2A кодирует белок в 1546 аминокислот, который содержит домены эпидермального фактора роста ламинина и фибронектина типа III. Эти домены обнаруживали и в других белковых компонентах базальной пластинки, внеклеточного матрикса и в адгезивных молекулах клеток. В исследовании дана характеристика интрон-экзонной структуры гена, состоящего из 21 экзона. В гене было обнаружено три мутации (239=242insCGTA, R334W, Т1515М) и описано 12 полиморфных вариантов у больных с легкими и тяжелыми формами синдрома Ушера типа II с прогрессирующим течением и сенсорно-нейрональной потерей слуха. При дальнейшем исследовании этого гена выявлено 15 новых мутаций у 57 неродственных пробандов, в том числе обнаружено три новые миссенс-мутации (C319Y, N346H и C419F). Установлено, что 58 независимых аллелей гена USH2A из 114 содержат патологические мутации, из которых наиболее часто (у 16 % больных) наблюдаемая — 2299delG, является мажорной.
- В районе короткого плеча хромосомы 3(р24.2-р23), в области маркеров D3S1578, D3S3647 и D3S3658, была картирована вторая форма второго типа USH2B.
В двух больших семьях с синдромом Ушера типа II с легкой формой пигментного ретинита не выявлено сцепления ни с маркерами 1q41, ни с хромосомой 3q25, т.е. у членов этих семей гены USH2A и USH3 не являлись причиной заболевания. Однако в дальнейшем было установлено, что при фенотипе синдрома Ушера типа II со слабовыраженными офтальмоскопическими проявлениями пигментного ретинита, который не был диагностирован до третьей декады жизни, имелось сцепление с локусом D5S484 на хромосоме 5q. Анализ гаплотипов длинного плеча хромосомы 5q указывает на то, что новый локус расположен между D5S428 и D5S433. К настоящему времени выделено девять таких семей.
В 1996 г. было обследовано 32 семьи с синдромом Ушера типа III (USH3) из географического изолята в Финляндии. Анализ сцепления позволил локализовать заболевание в длинном плече хромосомы 3(q21- q25) между маркерами D3S1299 и D3S3625, расстояние между которыми составляет около 1 сМ (сантиморган).
 По-видимому, существует еще и митохондриальная форма синдрома Ушера. Мутация С12258А в митохондриальном гене MTTS2 явилась причиной развития пигментного ретинита в сочетании с сенсорно-нейрональной формой потери слуха. Работы по картированию генов пигментного ретинита и синдромальных форм продолжаются.
По-видимому, существует еще и митохондриальная форма синдрома Ушера. Мутация С12258А в митохондриальном гене MTTS2 явилась причиной развития пигментного ретинита в сочетании с сенсорно-нейрональной формой потери слуха. Работы по картированию генов пигментного ретинита и синдромальных форм продолжаются.
В семьях, где имеются дети с синдромом Ушера, риск рождения больного ребенка составляет 25 %.
Различают 4 типа синдрома Ушера:
- тип I — пигментный ретинит и тотальная (абсолютная) глухота при отсутствии вестибулярных функций;
- тип II — пигментный ретинит, частичная глухота и интактная вестибулярная функция;
- тип III — пигментный ретинит, полная глухота, вестибулярная атаксия и в отдельных случаях психоз (синдром Халльгрена);
- тип IV — пигментный ретинит, тотальная глухота и задержка умственного развития.
Другая вариация классификации представляется следующим образом:
- грубая ретинопатия, сопровождающаяся функциональными изменениями,
- глубокая врожденная медленно прогрессирующая сенсорно-нейрональная потеря слуха,
- присутствие или отсутствие вестибулярного ответа на тепловой раздражитель,
- статус спиноцеребеллярный и высшей церебральной функции.
В зависимости от возраста появления симптомов ночной слепоты выделяют два типа синдрома Ушера
- Первый тип характеризуется затруднениями при передвижении в условиях слабой освещенности, возникающими в возрасте 5-6 лет,
- при втором типе нарушение ночного зрения возникает в 12-15 лет.
Острота зрения изменяется так же, как при пигментном ретините. При пигментации в макулярной области острота зрения понижается. Прогрессирующий тип заболевания часто наблюдается у больных с синдромом Ушера, которые имеют хорошее зрение. Изменения пигментного эпителия сетчатки схожи с таковыми при макулярной дистрофии типа «бычий глаз» и гипопигментации макулярной области, сочетающейся с пигментным ретинитом.
В последние годы с помощью компьютерной томографии было показано, что у больных с синдромом Ушера с глубокой глухотой имеет место мозжечковая (церебеллярная) атрофия затылочной доли, cavum septum pellucidum, cavum vergae или снижение церебеллярной циркуляции. Картина магнитного резонанса показывает патологически высокий сигнал, свидетельствующий об интенсивности поражения среднего мозга и мозжечка. Однако выявленные изменения мало связаны с клиническими симптомами синдрома Ушера.
В исследованиях, проведенных в последнее десятилетие, установлено нарушение регенерации кинетики зрительного пигмента в фовеа при нормальной световой чувствительности и сохранной остроте зрения.
Лечения синдрома нет. Однако с пациентами должны заниматься социальные работники, нейропсихиатры и воспитатели, используя технику, приспособленную для преподавания по индивидуальным программам. В семьях с близкородственными браками, в которых имеются больные с синдромом Ушера, высок риск рождения других больных детей. Однако определение носителей патологического гена все еще затруднено, хотя в последнее временя совокупность наследственных признаков (сцепление) и достижения молекулярной генетики вселяют надежду.
Источник