Синдром ушера 1 и 2 типа

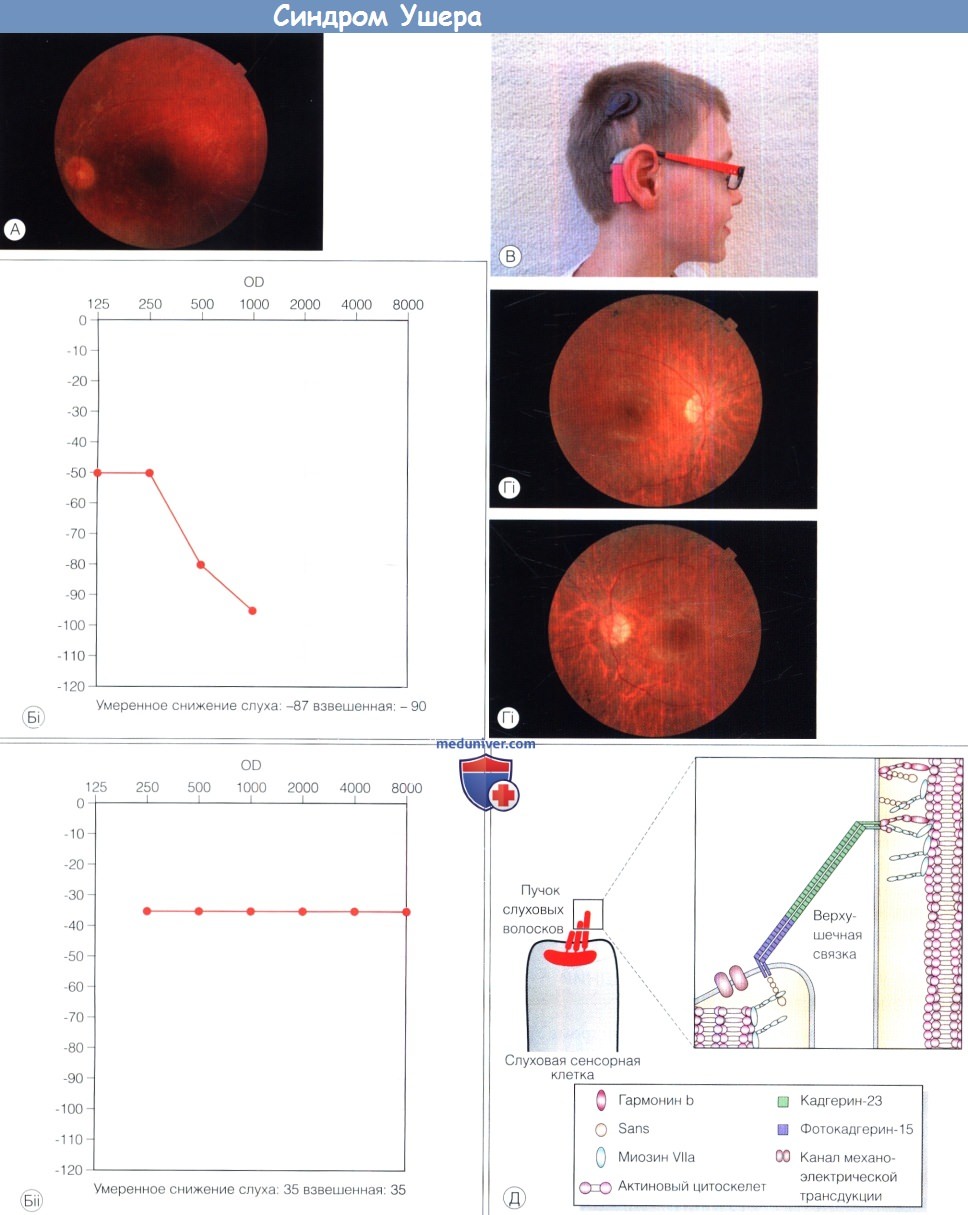
Синдром Ушера (Usher’s syndrome, USH) — глухой слепнущий ребенокСиндром Ушера (Usher’s syndrome, USH) — комбинация сенсоневральной глухоты с прогрессирующей дегенерацией сетчатки. Синдромом Ушера страдает один из каждых десяти глухих детей (5% всех случаев врожденной глухоты). Это наиболее часто встречающийся наследственный синдром, сопровождающийся глухотой и наиболее часто встречающийся синдром среди слепоглухонемых. Синдром подразделяется на три группы: синдром Ушера 1 типа (USH1), 2 типа (USH2) и 3 типа (USH3). Синдром Ушера 1 и 2 типов обычно диагностируется в раннем или старшем детском возрасте, соответственно, но может диагностироваться и позже, поскольку ухудшение зрения у глухого ребенка может оставаться незамеченным. Вестибулярная дисфункция связана с классической картиной синдрома Ушера 1 типа. Каждый тип синдрома Ушера наследуется по аутосомно-рецессивному механизму, но наблюдается генетическая гетерогенность этих заболеваний. Для каждого типа идентифицировано несколько генов. С помощью молекулярных исследований выявлено клиническое частичное совпадение классических первого и второго типов синдрома Ушера. Некоторые гены Ушера могут вызывать развитие изолированных глухоты или пигментного ретинита (USH2A). Таким детям настоятельно рекомендуется применение кохлеарных имплантов (особенно при USH1). Лечение дегенерации сетчатки все еще не разработано. а) Синдром Ушера 1 типа: наиболее тяжелая форма. Синдром Ушера 1 типа проявляется у детей тяжелой или полной сенсоневральной глухотой, ребенок поздно начинает садиться и ходить вследствие нарушения вестибулярной функции. В раннем детстве развивается дегенерация сетчатки. Уже в возрасте 2-3 лет изменения ЭРГ подтверждают диагноз дегенерации сетчатки, хотя глазное дно выглядит нормальным. Известно пять генов, мутации которых вызывают развитие заболевания, их частота варьирует в зависимости от исследуемой популяции: MY07A (считается основным геном, вызывающим синдром Ушера 1 типа, его мутации выявляются в половине случаев; также мутации этого гена вызывают несиндромальную глухоту), USH1C, CDH23, PCDH15 и USH1G. Хотя заболевание всегда протекает тяжело, его течение может различаться в зависимости от типа мутации. б) Синдром Ушера 2 типа: более позднее развитие дегенерации сетчатки у глухого ребенка. Врожденная сенсоневральная стабильная глухота от легкой до тяжелой степени тяжести, выраженные нарушения в области высоких частот и нормальная вестибулярная функция — основные признаки синдрома Ушера 2 типа. Дегенерация сетчатки развивается позже, чем при синдроме Ушера 1 типа, обычно в пубертатном периоде или позже. Синдром Ушера 2 типа и синдром Ушера 3 типа обычно не проявляются в течение первого десятилетия жизни и первоначально не вызывают развития изменений глазного дна. У многих пациентов сохраняется хорошая острота зрения, несмотря на сужение полей зрения. На ЭРГ на ранних стадиях изменения могут отсутствовать; однако изменения ЭРГ могут выявляться у детей и до развития клинической картины. Во втором десятилетии жизни болезнь проявляется ночной слепотой и утратой периферического зрения и необратимо прогрессирует. Идентифицировано три гена: USH2A (при изолированном пигментном ретините также выявлен мутантный ушерин), GPR98 и DFNB31. в) Синдром Ушера 3 типа: более поздний дебют, течение вариабельно. Возраст дебюта пигментного ретинита при синдроме Ушера 3 типа вариабелен, изменения сетчатки часто выявляются в возрасте старше двадцати лет, но могут быть ошибочно приняты за проявления двух других типов заболевания. Сенсорная глухота развивается уже постлингвально (в отличие от прелингвального развития глухоты при синдроме Ушера 1 и 2 типов), речь у таких пациентов развита нормально, но болезнь прогрессирует и приводит к полной слепоте. Вестибулярная дисфункция различной тяжести развивается у половины пациентов. Ген синдрома Ушера 3 типа особенно часто выявлялся у евреев-ашкенази и финнов. У пациентов с синдромом Ушера 1 и 2 типа также обнаружен мутантный USH3. г) Патогенез синдрома Ушера. В изучении патогенеза этого заболевания достигнут прогресс, установлено, что поражаются слуховые волоски внутреннего уха и фоторецепторные клетки сетчатки. Продукты гена USH1 формируют сеть, интерактом Ушера, который играет ключевую роль в раннем развитии пучков слуховых волосков. Они также являются ключевым компонентом механизма механоэлектрической трансдукции, необходимого для реализации функции слуха. В сетчатке интерактом Ушера действует на уровне цилиарной/пери-цилиарной зоны, в области соединения соединительного волоска с фоторецепторной клеткой. Эти структуры играю важную роль в транспорте протеинов между наружными и внутренними сегментами. Дефекты протеинов USH вызывают рано дебютирующую дисфункцию как внутреннего уха, так и сетчатки.
— Вернуться в содержание раздела «офтальмология» на сайте Оглавление темы «Наследственная патология сетчатки.»:
|
Источник
Синдром Ушера – редкое генетическое заболевание, характеризующееся глухотой из-за нарушения способности внутреннего уха и слуховых нервов передавать сенсорный (звуковой) сигнал в мозг (сенсорная потеря слуха).
Сопровождается пигментной ретинитой, расстройством, поражающим сетчатку и вызывающим прогрессирующую потерю зрения. Определено три клинических типа синдрома. Их отличают возраст появления симптомов и тяжесть. Usher наследуется как аутосомно-рецессивный генетический признак.
Введение
Впервые описан в 1858 году Альбрехтом фон Графе, назван в честь Чарльза Ушера, шотландского офтальмолога, идентифицировавшего наследственный, рецессивный характер наследования заболевания.

Признаки
Характеризуется глухотой из-за нарушения способности внутреннего уха и слуховых нервов передавать сенсорный (звуковой) вход в мозг (сенсорную потерю слуха). Также, аномальное скопление цветного (пигментного) материала на сетчатке (пигментный ретинит или РП).
РП вызывает дегенерацию сетчатки, приводящую к постепенной потере зрения и полной слепоте.
Сенсинологическая глухота нерва бывает глубокой, легкой, прогрессивной. Потеря зрения, вызванная пигментным ретинитом, начинается в детстве или позже в течение жизни. Часто впервые проявляется как плохое видение ночью или при низкой освещенности («ночная слепота»).
Исследования показывают, что четкое центральное зрение поддерживается в течение многих лет, даже когда боковое (периферическое) уменьшается.
Типы болезни
- Синдром Ушера типа 1 характеризуется глубокой потерей слуха при рождении (врожденная глухота), так и проблемами с балансом. Во многих случаях пострадавшие дети не умеют ходить до 18 месяцев или позже.
Проблемы со зрением начинаются в раннем подростковом возрасте, около десяти лет или немного раньше.
- Тип 2 характеризуется умеренной, иногда тяжелой потерей слуха в обоих ушах при рождении. Потеря слуха ухудшается с течением времени. Начало ночной слепоты происходит во время позднего подросткового возраста или начала двадцати лет.
Потери периферического зрения продолжаются, но центральное обычно сохраняется во взрослую жизнь. Визуальные проблемы имеют тенденцию прогрессировать медленнее, чем при типе 1.
- Тип 3 характеризуется более поздними нарушениями слуха, вариабельным балансом (вестибулярной) дисфункцией и РП, появляющейся между вторым и четвертым десятилетиями жизни.
Вестибулярные нарушения встречаются примерно у 50% детей с синдромом Ушера типа 3.
| 1 | 2 | 3 | |
| Слух | Глубокая потеря слуха или глухота при рождении. | От умеренной до тяжелой потери слуха при рождении. | Прогрессивная потеря в детстве или раннем подростковом возрасте. |
| Зрение | Снижение ночного видения к возрасту 10, прогрессирование к серьезной потери зрения на середине жизни. | Снижение ночного видения в подростковом возрасте, прогрессирование к серьезным потерям зрения на середине жизни. | Изменяется степень тяжести и возраста начала; проблемы ночного видения часто начинаются в подростковом возрасте и прогрессируют до тяжелой потери зрения в середине жизни. |
| Баланс (вестибулярная функция) | Проблемы с балансом с рождения. | Нормальный баланс. | От нормального до почти нормального равновесия в детстве; вероятность более поздних проблем. |

Почему возникает
Синдром ушера вызван мутациями определенных генов. До сих пор он ассоциировался с мутациями по меньшей мере в десяти:
- Тип 1: MYO7A (USH1B), USH1C, CDH23, PCDH15 (USH1F), SANS (USH1G), возможно, CIB2;
- 2 тип: USH2A, ADGRV1 (ранее называемый VLGR1), WHRN (DFNB31);
- Вариант 3: USH3A (CLRN1), HARS.
Эти гены предоставляют инструкции для нормального слуха, зрения, равновесия. Некоторые из них помогают специализированным клеткам, называемым волосковыми, передавать звук от внутреннего уха до мозга, ощущать свет и цвет сетчатке глаза. Функция некоторых белков, связанных с расстройством неизвестна.
Некоторые люди не имеют мутаций ни в одном из этих генов, поэтому, существуют другие, связанные с синдромом, которые еще не были идентифицированы.

Все типы синдрома Ушера наследуются как аутосомно-рецессивные черты. Большинство генетических заболеваний определяются статусом двух копий гена, полученных от отца и матери.
Когда наследуется один ген заболевания и один нормальный, то человек становится носителем без проявления симптомов.
- Риск того, что оба родителя-носителя передадут измененный ген детям – 25%.
- Риск иметь ребенка, который является носителем, таким как родители, составляет 50%.
- Шанс на получение ребенком нормальных генов от обоих родителей – 25%.
Риск одинаковый для мужчин и женщин.
Родители, близкие родственники имеют более высокий шанс, чем неродственные связи. Ненормальный ген, увеличивает риск иметь детей с рецессивным генетическим расстройством.
Затронутые популяции
Синдром Usher затрагивает три – десять из 100 000 человек во всем мире. Среди людей еврейской национальности Израиля, Берлина, Германии было обнаружено более высокое, чем среднее число пострадавших.
Часто поражаются Французские канадцы Луизианы, Аргентинцы испанского происхождения, нигерийские африканцы.
Тип 3, самая редкая форма в большинстве популяций, составляет около 40% пациентов Финляндии. Это самое распространенное генетическое заболевание, связанное с потерями зрения, слуха. Примерно 10 процентов всех случаев умеренной и глубокой глухоты у детей случается из-за типа 1 и 2.

Связанные нарушения
Симптомы следующих расстройств бывают сходны, сравнения полезны для постановки дифференциального диагноза:
Синдром Alström
Наследственное расстройство, характеризующееся дегенерацией сетчатки с нистагмом и потерей центрального зрения. Заболевание связано с ожирением в детстве. Сенсинологическая глухота и сахарный диабет развиваются после десятилетнего возраста.
Краснуха (немецкая корь)
Острое вирусное расстройство, вызывающее беспокойство при болезни на первых трех месяцев беременности, поскольку может вызвать отклонения плода. Эти аномалии включают нарушения слуха, зрения, пороки развития у ребенка.
Пигментный ретинит (RP)
Содержит большую группу наследственных нарушений зрения, вызывающих прогрессирующую дегенерацию сетчатки.
Периферическое (боковое) зрение постепенно уменьшается и теряется в большинстве случаев.
Центральное зрение обычно остается сохранно до позднего возраста. Некоторые формы RP связаны с глухотой, ожирением, заболеваниями почек, другими проблемами здоровья, включая поражения центральной нервной системы, нарушения обмена веществ, хромосомные аномалии
Диагностика
Синдром Ушера диагностируется при помощи обследования слуха, координации движения и зрения. При слуховом (аудиологическом) осмотре измеряется частота и громкость звуков, которые слышит человек.

Электроретинограмма измеряет электрический ответ на светочувствительных клетках сетчатки глаза. Осмотр сетчатки проводится для наблюдения за структурами задней части глаза.
Вестибулярную (балансовую) функцию оценивают с помощью тестов на разные части системы. Генетическое тестирование доступно для большинства генов, связанных с расстройством.

Лечение
Лечение синдрома Ушера направлено на конкретные симптомы. Оно требует скоординированных усилий группы медицинских специалистов, таких как педиатры, терапевты, отоларингологи, аудиологи, офтальмологи и другие специалисты здравоохранения.
Следует оценивать чувствительность органов дыхания. Возможности коммуникации изучаются как можно раньше, чтобы обеспечить ребенку прочную языковую базу. Слуховые аппараты или кохлеарные имплантаты принесут пользу большинству младенцев и детей с синдромом Ушера.
Язык жестов можно изучить как вариант связи. Люди, которые теряют зрение, изучают тактильные знаки. Раннее вмешательство важно для того, чтобы дети с синдромом Ушера достигли своего потенциала. Для детей с сенсоневральной глухотой или глухонемой требуются дополнительные медицинские, социальные, профессиональные услуги.
В настоящее время нет лечения расстройства, хотя исследователи работают над генетической и другой терапией для восстановления потери зрения, слуха.
Терапия витаминами
Исследования показали, для людей с типичным синдромом Ушера и типом 2, прием определенной суточной дозы витамина А замедляет дегенерацию сетчатки. Эксперты рекомендуют, чтобы взрослые пациенты с общими формами расстройства принимали витамин А пальмитат 15 000 МЕ ежедневно под наблюдением офтальмологов. Соблюдали регулярную сбалансированную диету и избегали приема витамина Е высоких доз.
Поскольку долгосрочная добавка витамина А высокой дозой (превышающая 25 000 МЕ) вызывает побочные эффекты, такие как болезни печени. Поэтому пациентам необходимо регулярно контролироваться врачами при приеме таких добавок.
Лица с пигментным ретинитом в сочетании с синдромом Ушера могут рассчитывать на помощь офтальмологов по коррекции слабого зрения. Другое лечение является симптоматическим и поддерживающим.
Генетическая консультация рекомендуется для пострадавших лиц и их семей.
Понравилась статья? Поделись с друзьями:
Источник
 Синдром Ашера (Ушера, англ. Usher syndrome, врожденная нейросенсорная глухота и пигментный ретинит) представляет собой аутосомно-рецессивное заболевание, включающее в себя зрительные (пигментный ретинит) и слуховые/вестибулярные нарушения. Распространенность синдрома Ашера варьирует от 4 до 17 на 100 000 человек, представлен тремя клиническими подтипами: USH1, USH2 и USH3, которые различаются степенью тяжести нарушений слуха, наличием или отсутствием вестибулярной дисфункции и началом развития пигментного ретинита.
Синдром Ашера (Ушера, англ. Usher syndrome, врожденная нейросенсорная глухота и пигментный ретинит) представляет собой аутосомно-рецессивное заболевание, включающее в себя зрительные (пигментный ретинит) и слуховые/вестибулярные нарушения. Распространенность синдрома Ашера варьирует от 4 до 17 на 100 000 человек, представлен тремя клиническими подтипами: USH1, USH2 и USH3, которые различаются степенью тяжести нарушений слуха, наличием или отсутствием вестибулярной дисфункции и началом развития пигментного ретинита.
Еще в 1858 г. Von Graefe сообщил о 5 случаях пигментного ретинита с тугоухостью с предполагаемой рецессивной формой наследования. Синдром Ушера — редко наблюдаемое заболевание; в общей популяции он встречается с частотой 3 случая на 100 000. Этот синдром диагностируют у 5-8 % больных с наследственной глухотой. Поскольку частота носительства мутантного гена составляет 1 на 100 человек при рецессивном типе наследования, этот синдром манифестирует редко. Точность диагностики возросла с появлением квантитативной аудиометрии. Истинный синдром Ушера характеризуется грубой наследственной сенсорно-нейрональной стабильной (возможно, прогрессирующей) потерей слуха в сочетании с пигментной ретинопатией, не отличимой от пигментного ретинита, которая сопровождается значительным затруднением речи.
Характерными симптомами синдрома Ушера являются ночная слепота и сужение полей зрения, возникающие в возрасте от 5 до 15 лет. Некоторые авторы считают синдром Ушера генетически гетерогенным, другие — фено-типическим проявлением пигментного ретинита. Предполагают, что среди патогенетических механизмов этого заболевания, передаваемого по аутосомно-рецессивному типу, имеется множество трофических нарушений, которые не могут быть определены одним геном. Однако возникновение глазных симптомов связывают с мутацией гена родопсина. Анализ сцепления указывает на локализацию дефекта в длинном плече хромосомы 4 в области 4(q11- q13).
 В настоящее время синдром Ушера — группа генетически гетерогенных заболеваний, наследуемых аутосомно-рецессивно. По клиническим проявлениям выделяют три основных типа заболевания, причем первые два типа включают несколько форм.
В настоящее время синдром Ушера — группа генетически гетерогенных заболеваний, наследуемых аутосомно-рецессивно. По клиническим проявлениям выделяют три основных типа заболевания, причем первые два типа включают несколько форм.
- Первый тип синдрома Ушера представлен 6 формами.
- При анализе сцепления было показано, что первая форма, названная USH1A, локализуется в районе длинного плеча хромосомы 14 (q32.1- q32.3) между локусами D14S78 и D14S250, но ген пока не клонирован.
- Вторая форма, названная USH1B, является наиболее распространенной: ее диагностируют примерно у 75 % от общего количества больных с первым типом заболевания. Эту форму удалось локализовать на длинном плече хромосомы 11q13.5, в районе ДНК-маркера DUS533. В этом же районе был клонирован один из генов миозина типа VILA. Скрининг этого гена на возможные мутации показал их наличие, основная масса которых приводила к преждевременному терминированию синтеза белка.
- Третья форма первого типа — USH1C была картирована в районе короткого плеча хромосомы 11р15.1 и связана с маркером D11S419. В гене-кандидате, названном гармонином, были обнаружены мутации, приводящие к синтезу укороченного белка, в котором отсутствовали его основные домены.
- USH1D удалось картировать в районе длинного плеча хромосомы 10 между локусами D10S529 и D10S573.
- USH1E — сцепление с ДНК-маркерами D21S1905 и D21S1913, расположенными в длинном плече хромосомы 21q21. Генетическое расстояние между этими маркерами составляет около 15 сМ (сантиморган).
- USH1F, с помощью геномного скрининга была картирована на хромосоме 10 в районе локусов D10S199 и D10S596.
- Второй тип синдрома Ушера включает 2 формы.
- Первая из них — USH2A была картирована в районе длинного плеча хромосомы 1q41 между ДНК-маркерами D1S237 и D1S229. Был клонирован ген-кандидат, названный ушерином. Ген USH2A кодирует белок в 1546 аминокислот, который содержит домены эпидермального фактора роста ламинина и фибронектина типа III. Эти домены обнаруживали и в других белковых компонентах базальной пластинки, внеклеточного матрикса и в адгезивных молекулах клеток. В исследовании дана характеристика интрон-экзонной структуры гена, состоящего из 21 экзона. В гене было обнаружено три мутации (239=242insCGTA, R334W, Т1515М) и описано 12 полиморфных вариантов у больных с легкими и тяжелыми формами синдрома Ушера типа II с прогрессирующим течением и сенсорно-нейрональной потерей слуха. При дальнейшем исследовании этого гена выявлено 15 новых мутаций у 57 неродственных пробандов, в том числе обнаружено три новые миссенс-мутации (C319Y, N346H и C419F). Установлено, что 58 независимых аллелей гена USH2A из 114 содержат патологические мутации, из которых наиболее часто (у 16 % больных) наблюдаемая — 2299delG, является мажорной.
- В районе короткого плеча хромосомы 3(р24.2-р23), в области маркеров D3S1578, D3S3647 и D3S3658, была картирована вторая форма второго типа USH2B.
В двух больших семьях с синдромом Ушера типа II с легкой формой пигментного ретинита не выявлено сцепления ни с маркерами 1q41, ни с хромосомой 3q25, т.е. у членов этих семей гены USH2A и USH3 не являлись причиной заболевания. Однако в дальнейшем было установлено, что при фенотипе синдрома Ушера типа II со слабовыраженными офтальмоскопическими проявлениями пигментного ретинита, который не был диагностирован до третьей декады жизни, имелось сцепление с локусом D5S484 на хромосоме 5q. Анализ гаплотипов длинного плеча хромосомы 5q указывает на то, что новый локус расположен между D5S428 и D5S433. К настоящему времени выделено девять таких семей.
В 1996 г. было обследовано 32 семьи с синдромом Ушера типа III (USH3) из географического изолята в Финляндии. Анализ сцепления позволил локализовать заболевание в длинном плече хромосомы 3(q21- q25) между маркерами D3S1299 и D3S3625, расстояние между которыми составляет около 1 сМ (сантиморган).
 По-видимому, существует еще и митохондриальная форма синдрома Ушера. Мутация С12258А в митохондриальном гене MTTS2 явилась причиной развития пигментного ретинита в сочетании с сенсорно-нейрональной формой потери слуха. Работы по картированию генов пигментного ретинита и синдромальных форм продолжаются.
По-видимому, существует еще и митохондриальная форма синдрома Ушера. Мутация С12258А в митохондриальном гене MTTS2 явилась причиной развития пигментного ретинита в сочетании с сенсорно-нейрональной формой потери слуха. Работы по картированию генов пигментного ретинита и синдромальных форм продолжаются.
В семьях, где имеются дети с синдромом Ушера, риск рождения больного ребенка составляет 25 %.
Различают 4 типа синдрома Ушера:
- тип I — пигментный ретинит и тотальная (абсолютная) глухота при отсутствии вестибулярных функций;
- тип II — пигментный ретинит, частичная глухота и интактная вестибулярная функция;
- тип III — пигментный ретинит, полная глухота, вестибулярная атаксия и в отдельных случаях психоз (синдром Халльгрена);
- тип IV — пигментный ретинит, тотальная глухота и задержка умственного развития.
Другая вариация классификации представляется следующим образом:
- грубая ретинопатия, сопровождающаяся функциональными изменениями,
- глубокая врожденная медленно прогрессирующая сенсорно-нейрональная потеря слуха,
- присутствие или отсутствие вестибулярного ответа на тепловой раздражитель,
- статус спиноцеребеллярный и высшей церебральной функции.
В зависимости от возраста появления симптомов ночной слепоты выделяют два типа синдрома Ушера
- Первый тип характеризуется затруднениями при передвижении в условиях слабой освещенности, возникающими в возрасте 5-6 лет,
- при втором типе нарушение ночного зрения возникает в 12-15 лет.
Острота зрения изменяется так же, как при пигментном ретините. При пигментации в макулярной области острота зрения понижается. Прогрессирующий тип заболевания часто наблюдается у больных с синдромом Ушера, которые имеют хорошее зрение. Изменения пигментного эпителия сетчатки схожи с таковыми при макулярной дистрофии типа «бычий глаз» и гипопигментации макулярной области, сочетающейся с пигментным ретинитом.
В последние годы с помощью компьютерной томографии было показано, что у больных с синдромом Ушера с глубокой глухотой имеет место мозжечковая (церебеллярная) атрофия затылочной доли, cavum septum pellucidum, cavum vergae или снижение церебеллярной циркуляции. Картина магнитного резонанса показывает патологически высокий сигнал, свидетельствующий об интенсивности поражения среднего мозга и мозжечка. Однако выявленные изменения мало связаны с клиническими симптомами синдрома Ушера.
В исследованиях, проведенных в последнее десятилетие, установлено нарушение регенерации кинетики зрительного пигмента в фовеа при нормальной световой чувствительности и сохранной остроте зрения.
Лечения синдрома нет. Однако с пациентами должны заниматься социальные работники, нейропсихиатры и воспитатели, используя технику, приспособленную для преподавания по индивидуальным программам. В семьях с близкородственными браками, в которых имеются больные с синдромом Ушера, высок риск рождения других больных детей. Однако определение носителей патологического гена все еще затруднено, хотя в последнее временя совокупность наследственных признаков (сцепление) и достижения молекулярной генетики вселяют надежду.
Источник