Синдром удлиненного интервала qt википедия


Синдром удлиненного интервала QT – генетически гетерогенное наследственное состояние, характеризующееся нарушением структуры и функциональности некоторых ионных каналов кардиомиоцитов. Выраженность проявлений патологии колеблется в очень широких пределах – от практически бессимптомного течения (выявляются только электрокардиологические признаки) до тяжелой глухоты, обмороков и аритмий. Определение синдрома удлиненного интервала QT производится на основании данных электрокардиологических исследований и молекулярно-генетических анализов. Лечение зависит от формы патологии и может включать в себя постоянный или курсовой прием бета-андреноблокаторов, препаратов магния и калия, а также установку дефибриллятора-кардиовертера.
Общие сведения
Синдром удлиненного интервала QT – группа кардиологических расстройств генетической природы, при которых нарушается прохождение ионных токов в кардиомиоцитах, что способно приводить к аритмиям, обморокам и внезапной сердечной смерти. Впервые подобное состояние было выявлено в 1957 году норвежскими врачами А. Джервеллом и Ф. Ланге-Нильсеном, которые описали сочетание у больного врожденной глухоты, синкопальных приступов и удлинения интервала QT. Несколько позже, в 1962-64 годах были выявлены схожие симптомы у пациентов, имеющих нормальный слух – такие случаи были описаны независимо друг от друга К. Романо и О. Уорд.
Это, а также дальнейшие открытия определили разделение синдрома удлиненного интервала QT на два клинических варианта – Романо-Уорда и Джервелла-Ланге-Нильсена. Первый наследуется по аутосомно-доминантному механизму, его частота в популяции составляет 1 случай на 5 000 населения. Встречаемость синдрома удлиненного интервала QT типа Джервелла-Ланге-Нильсена колеблется в пределах 1-6:1 000 000, он характеризуется аутосомно-доминантным путем наследования и более выраженными проявлениями. По некоторым данным, все формы синдрома удлиненного интервала QT ответственны за треть случаев внезапной сердечной смерти и около 20% внезапной младенческой смерти.

Синдром удлиненного интервала QT
Причины и классификация
В настоящее время удалось идентифицировать 12 генов, мутации в которых приводят к развитию синдрома удлиненного интервала QT, все они кодируют те или иные белки, входящие в состав ионных каналов кардиомиоцитов, отвечающих за натриевый или калиевый ионный ток. Удалось также найти причины различий в клиническом течении этого заболевания. Аутосомно-доминантный синдром Романо-Уорда обусловлен мутацией только одного гена и поэтому может протекать бессимптомно или, как минимум, с отсутствием нарушений слуха. При типе Джервелла-Ланге-Нильсена имеется дефект двух генов – этот вариант, помимо кардиологических симптомов, всегда сопровождается двухсторонней нейросенсорной глухотой. На сегодняшний день известно, мутации каких генов обуславливают развитие синдрома удлиненного интервала QT:
- Синдром удлиненного интервала QT тип 1 (LQT1) обусловлен мутацией гена KCNQ1, расположенного на 11-й хромосоме. Дефекты этого гена наиболее часто выявляются при наличии данного заболевания. Он кодирует последовательность альфа-субъединицы одной из разновидностей калиевых каналов кардиомиоцитов (lKs)
- Синдром удлиненного интервала QT тип 2 (LQT2) вызывается дефектами в гене KCNH2, который локализован на 7-й хромосоме и кодирует аминокислотную последовательность белка – альфа-субъединицы другого типа калиевых каналов (lKr).
- Синдром удлиненного интервала QT тип 3 (LQT3) обусловлен мутацией гена SCN5A, расположенного на 3-й хромосоме. В отличие от предыдущих вариантов патологии, при этом нарушается работа натриевых каналов кардиомиоцитов, так как данный ген кодирует последовательность альфа-субъединицы натриевого канала (lNa).
- Синдром удлиненного интервала QT тип 4 (LQT4) – достаточно редкий вариант состояния, вызванный мутацией гена ANK2, который расположен на 4-й хромосоме. Продуктом его экспрессии является белок анкирин В, который в организме человека участвует в стабилизации структуры микротрубочек миоцитов, а также выделяется в клетках нейроглии и сетчатки глаза.
- Синдром удлиненного интервала QT тип 5 (LQT5) – разновидность заболевания, которая обусловлена дефектом в гене KCNE1, локализованном на 21-й хромосоме. Он кодирует один из белков ионных каналов – бета-субъединицу калиевых каналов типа lKs.
- Синдром удлиненного интервала QT типа 6 (LQT6) вызывается мутацией в гене KCNE2, расположенного также на 21-й хромосоме. Продуктом его экспрессии является бета-субъединица калиевых каналов типа lKr.
- Синдром удлиненного интервала QT типа 7 (LQT7, другое название – синдром Андерсена, в честь педиатра Е. Д. Андерсена, описавшего это заболевание в 70-х годах) обусловлен дефектом гена KCNJ2, который локализуется на 17-й хромосоме. Как и в случае предыдущих вариантов патологии, этот ген кодирует одну из белковых цепей калиевых каналов.
- Синдром удлиненного интервала QT типа 8 (LQT8, другое название – синдром Тимоти, в честь К. Тимоти, описавшей это заболевание) вызван мутацией гена CACNA1C, который располагается на 12-й хромосоме. Этот ген кодирует альфа-1-субъединицу кальциевого канала L-типа.
- Синдром удлиненного интервала QT тип 9 (LQT9) обусловлен дефектом гена CAV3, локализованного на 3-й хромосоме. Продуктом его экспрессии является белок кавеолин 3, участвующий в формировании множества структур на поверхности кардиомиоцитов.
- Синдром удлиненного интервала QT тип 10 (LQT10) – причина этой разновидности заболевания кроется в мутации гена SCN4B, который располагается на 11-й хромосоме и отвечает за аминокислотную последовательность бета-субъединицы натриевых каналов.
- Синдром удлиненного интервала QT тип 11 (LQT11) вызывается дефектами в гене AKAP9, расположенном на 7-й хромосоме. Он кодирует специфический белок – А-киназу центросом и комплекса Гольджи. Функции этого протеина на сегодняшний день изучены недостаточно.
- Синдром удлиненного интервала QT тип 12 (LQT12) обусловлен мутацией гена SNTA1, локализованного на 20-й хромосоме. Он кодирует альфа-1-субъединицу белка синтрофина, участвующего в регуляции деятельности натриевых каналов кардиомиоцитов.
Несмотря на широкое генетическое разнообразие синдрома удлиненного интервала QT, общие звенья его патогенеза в целом одинаковы для каждой из форм. Данное заболевание относят к группе каналопатий из-за того, что его причиной выступают нарушения в строении тех или иных ионных каналов. В результате этого процессы реполяризации миокарда происходят неравномерно и не одновременно в различных частях желудочков, что становится причиной удлинения интервала QT. Кроме того, значительно возрастает чувствительность миокарда к влияниям симпатической нервной системы, что становится причиной частых тахиаритмий, способных приводить к жизнеугрожающим фибрилляциям желудочков. При этом у разных генетических типов синдрома удлиненного интервала QT отмечается различная чувствительность к тем или иным воздействиям. Например, LQT1 характеризуется синкопальными приступами и аритмией при физической нагрузке, при LQT2 аналогичные проявления наблюдаются при громких и резких звуках, для LQT3, напротив, более характерно развитие аритмий и фибрилляций в спокойном состоянии (например, во сне).
Симптомы удлиненного интервала QT
Проявления синдрома удлиненного интервала QT достаточно разнообразны. При более тяжелом клиническом типе Джервелла-Ланге-Нильсена у больных отмечается глухота, частые обмороки, головокружения, слабость. Кроме того, в ряде случаев при этом состоянии регистрируются эпилептоподобные судорожные припадки, что нередко приводит к неправильной диагностике и лечению. По данным некоторых врачей-генетиков, от 10 до 25% больных с синдромом удлиненного интервала QT получают неправильное лечение, и у них развивается внезапная сердечная или младенческая смерть. Возникновение тахиаритмий и синкопальных состояний зависит от внешних влияний – например, при LQT1 это может происходить на фоне физической нагрузки, при LQT2 потеря сознания и фибрилляция желудочков может возникать от резких и громких звуков.
Более легкая форма синдрома удлиненного интервала QT (тип Романо-Уорда) характеризуется преходящими синкопальными состояниями (обмороками) и редкими приступами тахиаритмии, однако нарушения слуха при этом отсутствуют. В ряде случаев подобная форма заболевания вообще ничем не проявляет себя, за исключением электрокардиографических данных, и является случайной находкой при медицинском обследовании. Тем не менее, даже при таком течении синдрома удлиненного интервала QT риск внезапной сердечной смерти из-за фибрилляции желудочков во много раз выше, нежели у здорового человека. Поэтому и эта разновидность патологии требует тщательного изучения и профилактического лечения.
Диагностика
Диагностика синдрома удлиненного интервала QT производится на основании изучения анамнеза больного, электрокардиологических и молекулярно-генетических исследований. При расспросе пациента часто обнаруживаются эпизоды обмороков, головокружений, ощущения сердцебиений, но при легких формах патологии их может и не быть. Иногда аналогичные проявления встречаются у кого-либо из родственников пациента, что указывает на семейный характер заболевания.
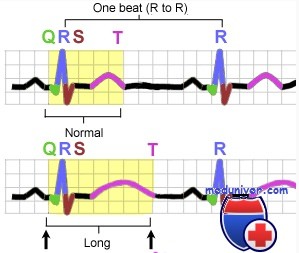
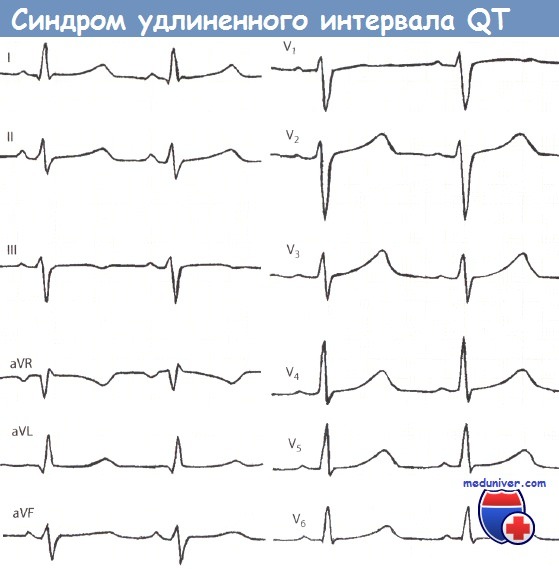
При любой форме синдрома удлиненного интервала QT будут выявляться изменения на ЭКГ – увеличение интервала QT до 0,6 секунд и более, возможно увеличение амплитуды зубца Т. Сочетание таких ЭКГ-признаков с врожденной глухотой говорит о наличии синдрома Джервелла-Ланге-Нильсена. Кроме того, часто необходимо холтеровское мониторирование работы сердца на протяжении суток для выявления возможных приступов тахиаритмий. Определение синдрома удлиненного интервала QT при помощи методов современной генетики на сегодняшний день возможно в отношении практических всех генетических типов этого заболевания.
Лечение синдрома удлиненного интервала QT
Терапия синдрома удлиненного интервала QT достаточно сложна, многие специалисты рекомендуют при этом заболевании одни схемы и отвергают другие, но какого-либо единого протокола лечения этой патологии не существует. Универсальными препаратами считаются бета-адреноблокаторы, которые уменьшают риск развития тахиаритмий и фибрилляций, а также снижают степень симпатических воздействий на миокард, но при LQT3 они малоэффективны. В случае синдрома удлиненного интервала QT типа 3 более разумно использовать антиаритмические препараты класса В1. Эти особенности лечения заболевания повышают потребность в молекулярно-генетической диагностике для определения типа патологии. В случае частых приступов тахиаритмий и высокого риска развития фибрилляции рекомендуется имплантация кардиостимулятора или дефибриллятора-кардиовертера.
Прогноз
Прогноз синдрома удлиненного интервала QT, по мнению большинства специалистов, неопределенный, так как это заболевание характеризуется широким спектром выраженности симптомов. Кроме того, отсутствие проявлений патологии, за исключением электрокардиографических данных, не гарантирует внезапного развития фатальной фибрилляции желудочков под воздействием внешних или внутренних факторов. При выявлении синдрома удлиненного интервала QT необходимо произвести тщательное кардиологическое обследование и генетическое определение типа заболевания. На основе полученных данных разрабатывается схема лечения, призванная снизить вероятность внезапной сердечной смерти, или принимается решение об имплантации кардиостимулятора.
Источник
Синдром удлиненного QT (LQT): причины, диагностика, лечениеЭтиология и встречаемость синдрома удлиненного QT. Синдромы удлиненного QT (LQT) — разнородная панэтническая группа нарушений, получивших название каналопатий, поскольку они вызываются дефектами в ионных каналах сердца. Распространеность синдромов удлиненного QT приблизительно 1 на 5000-7000 человек. Большинство случаев удлиненного QT вызвано мутациями в пяти известных генах ионных каналов сердца (KCNQ1, KCNH2, SCN5A, KCNE1.KKCNE2). Генетика, лежащая в основе синдромов удлиненного QT, сложна. Во-первых, существует локусная гетерогенность. Наиболее частый из синдромов удлиненного QT, аутосомно-доминантный синдром Романо-Уорда (MIM №192500), вызван преимущественно мутациями в двух локусах, KCNQ1 и KCNH2, а также содействующим третьим локусом, SCN5A. Во-вторых, разные мутантные аллели в одном и том же локусе могут вызывать два различающихся синдрома удлиненного QT, синдром Романо-Уорда и аутосомно-рецессивный синдром Джервелла-Ланге-Нильсена (MIM №220400). Патогенез синдрома удлиненного QTСиндром удлиненного QT вызывается дефектами реполяризации в клетках сердца. Реполяризация — управляемый процесс, требующий баланса между направленным внутрь клетки потоком натрия и кальция и из клетки — калия. Дисбаланс удлиняет или укорачивает длительность потенциала действия, вызывающего соответственно удлинение или сокращение интервала QT на электрокардиограмме. Большинство случаев синдрома удлиненного QT вызваны мутациями с утратой функции в генах, кодирующих субъединицы или полные белки каналов калия (названия этих генов начинаются с KCN). Эти мутации уменьшают реполяризацию, тем самым продлевая потенциал действия клетки и уменьшая порог для последующей деполяризации. У других пациентов с синдромом удлиненного QT мутации с усилением функции в гене натриевого канала, SCN5A, ведут к повышенному притоку натрия, вызывая аналогичные изменения потенциала действия и эффекты реполяризации.
Фенотип и развитие синдрома удлиненного QTСиндромы удлиненного QT характеризуются удлинением интервала QT и аномалиями зубца Т на электрокардиограмме, включая тахиаритмию и полиморфную желудочковую тахикардию. Желудочковая тахикардия характеризуется изменением амплитуды и скручиванием комплекса QRS. Полиморфная желудочковая тахикардия связана с удлиненным интервалом QT и обычно заканчивается спонтанно, но может упорствовать и прогрессировать в фибрилляцию желудочков. При самом частом варианте синдрома удлиненного QT, Романо-Уорда, обмороки из-за аритмии сердца — наиболее частый признак. Если ребенок остается недиагностированным или не получает лечение, синкопальные состояния повторяются и могут быть летальными в 10-15% случаев. Тем не менее от 30 до 50% индивидуумов с синдромом удлиненного QT никогда не имеют синкопальных симптомов. Сердечные эпизоды чаще всего встречаются в возрасте от 9 до 12 лет, уменьшаясь со временем. Эпизоды могут происходить в любом возрасте, если спровоцированы приемом медикаментов, удлиняющих интервал QT. Нефармакологические триггеры сердечных событий при синдроме Романо-Уорда отличаются в зависимости от ответственного гена. Триггеры LQT1 — обычно адренергические стимулы, включая физическую нагрузку и внезапные эмоции (испуг). Лица с LTQ2 находятся в риске как при нагрузке, так и в покое, а также при слуховых стимулах, например звонок будильника или телефона. Пациенты с LQT3 имеют эпизоды с замедлением сердечных показателей в периоды отдыха и сна. Кроме того, 40% случаев LQT1 проявляют себя до 10-летнего возраста; симптоматика появляется до 10 лет жизни только в 10% случаев LTQ2 и крайне редко при LQT3. Синдром LQT5 редкий, о течении и триггерах известно меньше. Синдром удлиненного QT имеет неполную пенетрантность, как с точки зрения электрокардиографических аномалий, так и синкопальных эпизодов. До 30% больных могут иметь интервалы QT, перекрывающиеся с нормальными колебаниями. Варьирующаяся экспрессия заболевания может происходить как внутри семьи, так и между семьями. Из-за неполной пенетрантности для точного диагноза у членов семьи часто необходима нагрузочная электрокардиография. Синдромы удлиненного QT могут сопровождаться дополнительными данными при медицинском осмотре. Например, синдром Джервелла-Ланге-Нильсена (MIM №220400) характеризуется глубокой врожденной нейросенсорной глухотой в сочетании с синдромом удлиненного QT. Это аутосомно-рецессивное заболевание, вызываемое также определенными мутациями в одном из двух генов (KCNQ1 и KCNE1), участвующих в развитии аутосомно-доминантного синдрома Романо-Уорда. Гетерозиготные родственники пациентов с синдромом Джервелла-Ланге-Нильсена не глухие, но имеют 25% риск развития синдрома удлиненного QT. Особенности фенотипических проявлений синдрома удлиненного QT:
Лечение синдрома удлиненного QTЛечение синдрома удлиненного QT направлено на предотвращение синкопальных эпизодов и остановки сердца. Оптимальное лечение зависит от идентификации ответственного в данном случае гена. Например, терапия b-адреноблокаторами до начала симптомов — наиболее эффективный метод при LQT1 и, отчасти, при LQT2, но его эффективность при LQT3 незначительна. При лечении b-адреноблокаторами необходимо тщательно проверять соответствие возрастным дозам, не прерывать прием лекарственных средств. Для больных с брадикардией могут оказаться необходимыми водители ритма; может потребоваться доступ к внешним дефибрилляторам. Имплантируемые кардиовертеры-дефибрилляторы могут быть необходимыми больным с LQT3 или некоторым лицам с синдромом удлиненного QT, для которых проблематична терапия бета-адреноблокаторами, например больным бронхиальной астмой, депрессией или сахарным диабетом, а также пациентам с остановкой сердца в анамнезе. Некоторые лекарства, например антидепрессивный препарат амитриптилин, фенилэфрин и дифенилгидрамин, или противогрибковые лекарства, включая флуконазол и кетоназол, должны быть исключены из-за их действия, удлиняющего интервал QT или повышения симпатико-тонии. Исключают также виды деятельности и спорта, связанные с интенсивной физической нагрузкой и эмоциональным стрессом.
Риски наследования синдрома удлиненного QTЛица с синдромом Романо-Уорда имеют 50% шанс родить ребенка с унаследованными мутациями в гене. Поскольку частота новых мутаций низкая, большинство больных имеют пораженного родителя (хотя, возможно, бессимптомного). Чрезвычайно важны и могут оказаться жизнесохраняющими подробный семейный анамнез и тщательная кардиологическая оценка членов семьи. Риск повторения для сибсов пациентов с синдромом Джервелла-Ланге-Нильсена — 25%, как и ожидается при аутосомно-рецессивном заболевании. Пенетрантность изолированного синдрома удлиненного QT без глухоты для гетерозиготных носителей синдрома Джервелла-Ланге-Нильсена — 25%. Пример синдрома удлиненного QT. А.Б., 30-летняя женщина с синдромом удлиненного QT (LQT), обратилась в генетическую клинику вместе с мужем, поскольку они планируют беременность. Пара хочет знать риск повторения этого заболевания у детей и подходящие методы генетического тестирования и пренатальной диагностики. Женщина также обеспокоена потенциальным влиянием беременности на ее собственное здоровье. Диагноз синдрома LQT установлен в начале третьего десятилетия жизни, когда она проходила обследование после внезапной смерти ее 15-летнего брата. В целом она — здоровый человек с нормальным слухом, отсутствием дисморфических признаков. У нее никогда не было обморочных состояний. Впоследствии электрокардиографические данные подтвердили диагноз синдрома у нее, ее отца и одной из теток по отцу. Молекулярное тестирование выявило миссенс-мутацию в гене KCNH2, ранее описанную в других семьях с синдромом Романо-Уорда, тип LQT2. Первоначально пациентка получала бета-адреноблокаторы, но ее кардиологи решили, что низкая эффективность b-адреноблокаторов при LQT2 и летальный случай у ее брата оправдывают имплантацию кардиовертера-дефибриллятора как ей, так и ее пораженным родственникам. Пациентка — первый человек в ее семье, проходящий генетическое консультирование по синдрому удлиненнго QT. — Также рекомендуем «Синдром Марфана: причины, диагностика, лечение» Оглавление темы «Наследственные болезни»:
|
Источник