Синдром супер мужчины что это

XYY-синдром, также известен как YY-синдром или Синдром Джейкобс (d)[1], — хромосомное заболевание, характерное только для мужчин. Носитель синдрома имеет дополнительную Y-хромосому, общий хромосомный набор составляет 44 аутосомы и три половые хромосомы. Внешне мужчины с дополнительной Y-хромосомой обычно не имеют существенных отличий от мужчин с обычным набором хромосом, но могут иметь ряд особенностей[2].
История исследования[править | править код]
Синдром был предсказан в работах Патрисии Джейкобс 1959 года по XXY-синдрому[1], а впервые обнаружен в 1961 году при случайном обследовании мужчины, дети которого имели ряд заболеваний (в частности, один из детей имел синдром Дауна)[3][4]. XYY-синдром был описан как так называемый «синдром сверх-самца» или «синдром сверх-мужчины» (англ. super-male syndrome), при этом носителям синдрома приписывалось агрессивное поведение и тенденция к криминальным действиям. Первые исследователи болезни в 1960-х годах обнаружили относительно высокое количество мужчин с этим синдромом среди обитателей тюрем и психиатрических клиник. Это послужило основой для стереотипа о «сверх-мужчине»[2].
Последующие исследования показали, что абсолютное большинство носителей синдрома никогда не имели отношения к преступности или психическим заболеваниям, но могут иметь повышенный риск проблем с обучением. Дополнительная Y-хромосома сама по себе не ведёт к чрезмерной агрессивности[2].
Генетические особенности[править | править код]
Диаграмма, показывающая формирование синдрома XYY. MI и MII являются стадиями мейоза, тогда как синие и розовые круги являются мужской и женской клетками соответственно, а синие и розовые полоски — Y- и X-хромосомы соответственно. Фиолетовая клетка имеет 2 Y-хромосомы и 1 Х-хромосому из-за слияния с мужской клеткой с 2 Y-хромосомами, что было связано с проблемами деления в MII мужчины
Причиной возникновения синдрома является нерасхождение Y-хромосом в анафазе II[источник не указан 2351 день] в процессе сперматогенеза. В результате появляется сперматозоид, несущий вторую Y-хромосому, в результате оплодотворения которым появляется ребёнок с 47 хромосомами в кариотипе (47 XYY). Синдром не является наследуемым состоянием, риск рождения второго ребёнка с этим синдромом не выше, чем в среднем в популяции. Частота появления — примерно 1 случай на 1000 мужчин. Большая часть носителей не знает о своей особенности[2].
Несмотря на наличие лишней хромосомы, сперма XYY-мужчины обычно несёт нормальный набор хромосом, ввиду того, что лишняя Y-хромосома элиминируется. Риск появления детей с хромосомными заболеваниями у большинства мужчин с XYY-синдромом не отличается от такого же риска у мужчины с нормальным генотипом. В то же время, число сперматозоидов с хромосомными аномалиями у некоторых мужчин с XYY-синдромом выше, но неизвестно, насколько существенно это повышает риск появления детей с хромосомными болезнями у таких отцов[2].
Фенотипические характеристики[править | править код]
Наличие второй Y-хромосомы в большинстве случаев не ведёт к каким-либо физическим отклонениям. В то же время, многие мужчины с XYY-синдромом имеют одну или несколько особенностей. При рождении они имеют нормальный рост, но часто быстрее растут в детстве. В среднем, во взрослом состоянии носитель выше, чем 75 % мужчин того же возраста. Некоторые мужчины с синдромом XYY имеют небольшие нарушения координации движений, в результате чего могут казаться неуклюжими. Фертильность чаще всего не нарушена, обычно такие мужчины гетеросексуальны и имеют нормальную сексуальную функцию. Тем не менее, описаны случаи существенного снижения фертильности, вплоть до бесплодия. У небольшого числа носителей также повышен уровень половых гормонов, связанных со сперматогенезом, что может вести к бесплодию ввиду нарушения образования спермы. Неизвестно, насколько высоко число случаев бесплодия у мужчин с XYY-синдромом. IQ находится в пределах нормы, но часто несколько ниже, чем у родных братьев и сестёр. Примерно половина носителей имеет проблемы с обучением, в частности, могут быть нарушения речи и чтения. Может быть повышен риск поведенческих проблем, таких как синдром гиперактивности, мужчины с XYY-синдромом часто импульсивны и эмоционально незрелы[2].
См. также[править | править код]
- Синдром 48, XYYY
- Синдром 49, XYYYY
- Синдром Клайнфельтера
- Синдром Шерешевского-Тёрнера
- Трисомия по Х-хромосоме
Примечания[править | править код]
Источник
Что такое XYY-синдром?
XYY–синдром (или также называемый как YY-синдром или Синдром Джейкобса), — это редкое хромосомное заболевание, которое поражает мужчин. Вызван синдром наличием дополнительной Y-хромосомы. Мужчины обычно имеют одну X и одну Y-хромосому. Тем не менее, люди с этим синдромом имеют одну Х и две Y-хромосомы.
По оценкам, болезнь затрагивает примерно 1 из 1000 живорожденных мальчиков.
Пострадавшие люди обычно очень высокие. Многие испытывают серьезные трудности с прыщами в подростковом возрасте. Дополнительные симптомы могут включать неспособность к обучению и поведенческие проблемы, такие как импульсивность. Интеллект обычно находится в нормальном диапазоне, хотя IQ в среднем на 10-15 баллов ниже, чем у братьев и сестер.
В прошлом было много неправильных представлений об этой болезни. Иногда его называли болезнью мужского пола, потому что считалось, что мужчины с этим синдромом чрезмерно агрессивны и им не хватает сочувствия. Недавние исследования показали, что это не так. Хотя люди с синдромом XYY имеют повышенный риск нарушения обучаемости и поведенческих проблем, они не слишком агрессивны и не подвержены повышенному риску какого-либо серьезного психического заболевания.
Поскольку больные мальчики подвержены более высокому риску нарушения обучаемости, им может быть полезна логопедия, репетиторство и общее понимание конкретных проблем, с которыми они сталкиваются. Хотя первые годы в школе могут быть более сложными для мальчиков с XYY-синдромом, они обычно ведут полноценную, здоровую и нормальную жизнь.
Признаки и симптомы XYY-синдрома

Клинические признаки XYY-синдрома часто неуловимы и не обязательно предполагают серьезное хромосомное расстройство. Соответственно, мужчинам с этим условием часто либо не диагностируют синдром, либо диагностируют неправильную патологию.
Наиболее распространенной физическим симптомом является увеличение роста, которое обычно проявляется после 5 или 6 лет и приводит к росту в среднем на около 6 футов и 3 дюйма в зрелом возрасте (см. фото выше).
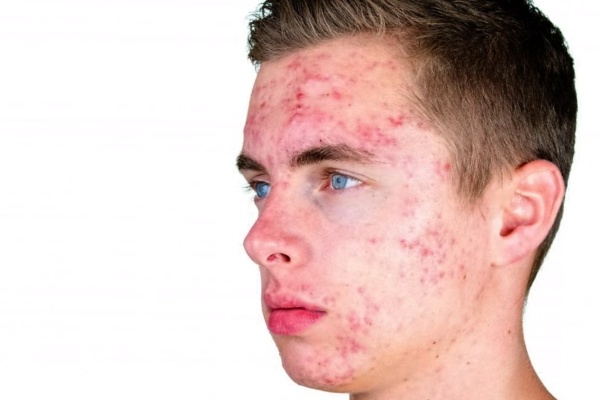
У некоторых людей с XYY-синдромом также развиваются тяжелые кистозные прыщи в подростковом возрасте. Рождаемость и половое развитие нормальны. Помимо возможности увеличения роста, большинство пострадавших людей обычно имеют нормальный внешний вид (фенотип).
Мальчики с синдромом Джейкобса обычно имеют нормальный интеллект, хотя в среднем IQ на 10–15 баллов ниже, чем у его братьев и сестер. У пострадавших мальчиков могут наблюдаться небольшие задержки в достижении основных этапов развития. Проблемы с обучением отмечались в 50% случаев, чаще всего с задержками речи и языковыми проблемами. Проблемы с чтением являются общими из-за повышенной частоты возникновения дислексии.
В некоторых случаях у затронутых людей развиваются поведенческие проблемы, такие как взрывной характер, гиперактивность, импульсивность, вызывающие действия или, в некоторых случаях, антиобщественное поведение.
Существует более высокий уровень дефицита внимания и гиперактивности и меньший повышенный риск развития расстройств аутистического спектра.
Причины XYY-синдрома
XYY-синдром — редкое хромосомное расстройство, вызванное наличием дополнительной Y-хромосомы. Обычно мужчины имеют 46 хромосом, включая одну Х и одну Y-хромосому. Мужчины с XYY-синдромом имеют 47 хромосом, две из которых являются Y-хромосомами.
Большинство случаев XYY-синдрома связаны с ошибкой клеточного деления в сперме до зачатия. Редко ошибка деления клеток возникает после зачатия, что приводит к появлению мозаики клеток с 46 хромосомами и 47 хромосомами.
Точная причина, по которой происходят эти ошибки в делении клеток, не понятна.
Похожие расстройства
Симптомы следующих расстройств могут быть похожи на симптомы XYY-синдрома. Сравнения могут быть полезны для дифференциальной диагностики:
Синдром Клайнфельтера связан с группой хромосомных нарушений у мужчин, в которых присутствует одна или несколько дополнительных Х-хромосом. Мужчины с классической формой расстройства имеют одну дополнительную Х-хромосому. У мужчин с различными формами синдрома Клайнфельтера имеются дополнительные Х и/или Y хромосомы. Дополнительная X и/или Y хромосома может влиять на физическое, развивающее, поведенческое и когнитивное функционирование.
Общие физические особенности могут включать высокий рост, отсутствие вторичного пубертатного развития, небольшие яички (гипогонадизм), замедленное пубертатное развитие и увеличение молочной железы (гинекомастия) в конце полового созревания. Эти особенности могут быть связаны с низким уровнем тестостерона и повышенным уровнем гонадотропина.
Синдром Сотоса (синдром церебрального гигантизма) — переменное генетическое заболевание, характеризующееся чрезмерным ростом до и после рождения. Одной из основных особенностей синдрома Сотоса является особый внешний вид лица, который включает в себя покраснение лица, ненормально выраженный лоб, наклоненные вниз веки, выступающую узкую челюсть, длинное узкое лицо и форму головы похожую на перевернутую грушу.
У большинства детей с синдромом Сотоса присутствуют задержки развития, которые могут включать двигательные и языковые задержки, а также умственную отсталость от легкой до тяжелой степени. Другие проблемы, связанные с синдромом Сотоса, включают желтуху у новорожденных, искривленный позвоночник (сколиоз), эпилепсию, косоглазие, кондуктивную потерю слуха, врожденные пороки сердца, почечные нарушения и поведенческие проблемы. Пострадавшие люди также имеют слегка повышенный риск развития определенных типов опухолей.
Синдром Сотоса вызван аномалией (мутацией) в гене NSD1.
Синдром Марфана — генетическое заболевание, поражающая соединительную ткань, которая является материалом между клетками организма, придающие тканям форму и силу. Соединительная ткань расположена по всему телу, и у людей с синдромом Марфана могут поражаться системы многих органов. Чаще всего поражаются сердечно-сосудистая система, скелетные и глазные системы.
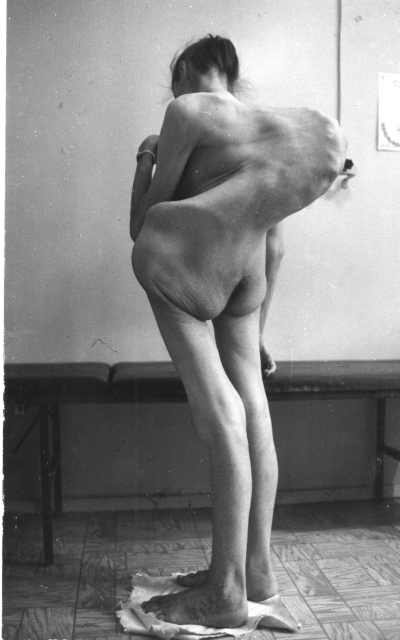
Основные симптомы включают чрезмерный рост костей рук и ног, ненормальное искривление позвоночника из стороны в сторону (сколиоз (см. фото)), вдавливание или выпячивание стенки грудной клетки, вывих линз глаз (смещение хрусталика глаза), близорукость, расширение (аневризма) и разрыв (расслоение) главной артерии, которая носит кровь от сердца (аорты), пролапс митрального клапана и обратный поток крови через аортальный и митральный клапаны (аортальная и митральная регургитация).
Специфические симптомы и степень выраженности синдрома Марфана сильно варьируются от случая к случаю. Синдром Марфана наследуется как аутосомно-доминантный признак.
С синдромом Марфана и связанными с ним расстройствами связаны дефекты или нарушения (мутации) гена фибриллина-1 (FBN1).
Диагностика XYY-синдрома
Диагноз XYY-синдрома ставится на основании тщательной клинической оценки, подробной истории болезни и специальных тестов (т. е. хромосомного анализа), которые выявляют наличие дополнительной Y-хромосомы (47, кариотип XYY).
Диагноз XYY-синдрома может быть поставлен до рождения (внутриутробно) с помощью амниоцентеза или взятия проб ворсин хориона. Во время амниоцентеза образец жидкости, который окружает развивающийся плод, удаляется и анализируется, в то время как взятия проб ворсин хориона включает в себя удаление образцов ткани из части плаценты. Хромосомные исследования, выполненные на таких образцах жидкости или тканях, могут выявить присутствие дополнительной Y-хромосомы.
— Клиническое тестирование и обследование
Оценка речи и языка должна проводиться в течение первых 24 месяцев. Оценка чтения должна проводиться в школьном возрасте, чтобы исключить дислексию. Поведенческая оценка должна рассматриваться для детей, которые испытывают трудности с такими симптомами, как импульсивность и плохое внимание.
Лечение XYY-синдрома
Лечение XYY-синдрома является симптоматическим и поддерживающим. Могут быть полезны логопедия, трудотерапия или помощь в обучении.
В большинстве случаев пострадавшие люди очень чутко реагируют на раннее вмешательство и лечение, и проблемы могут полностью решится в течение нескольких лет.
Лечение прыщей может помочь пострадавшему с самооценкой.
Дефицит внимания и гиперактивность, трудности с социальным взаимодействием или другие поведенческие проблемы можно лечить с помощью терапии или медикаментов так же, как у людей, у которых нет XYY.
Источник
Имеет место приблизительно у одного из 1000 мужчин. Обычно такие люди выше среднего роста, отличаются огромным количеством прыщей, минимальными нарушениями скелета. Раньше бытовало предположение, что «супермужчины» более агрессивны и развиваются не так, как люди с обычным генотипом. Однако это заключение оказалось преувеличением. Национальная Академия наук сделала вывод, что нет данных, свидетельствующих о наличии связи между лишними У-хромосомами и агрессивным, насильственным поведением.
1 20 Часть I. Начало
Окончание табл. 3.1
ХРОМОСОМНЫЕ СиндромТернера (ХО)
Имеет место приблизительно у одной из 10 000 женщин. Одна из Х-хромосом либо потеряна, либо неактивна. Женщины с синдромом Тернера обычно выглядят как незрелые женщины — у них не развиваются вторичные половые признаки. Также у них отсутствуют внутренние репродуктивные органы. Они могут обладать очень низким ростом и иногда — умственной отсталостью. Это расстройство обычно проявляется в пубертатном периоде. Гормонально-замещающая терапия может помочь привести внешний вид в большее соответствие с нормой.
Главные источники: OMIM, 2000; Pasternak, 1999.
Драматическим примером аномалии, обычно сцепленной с полом, является гемофилия,проявление которой более вероятно у мужчин, чем у женщин. Гемофилия представляет собой целую группу расстройств в том смысле, что дефекты различных генов Х-хромосомы (а в одной форме некоторой аутосомы) разрушают различные метаболические пути, которые отвечают за нормальную свертываемость крови. То есть в зависимости от того, какой ген поврежден, теряются один или несколько различных факторов плазмы крови. Однако каждая форма следует обычному доминантно-рецессивному паттерну.
Гемофилия — сравнительно редкое, но серьезное нарушение, от которого пока не найдено другого лечения, кроме регулярного переливания нормальной здоровой крови. У людей, страдающих гемофилией, кровь из малейшего пореза может течь неопределенно долго. Особенно опасны для них любые внутренние кровотечения, потому что они могут оставаться незамеченными и приводить к смертельному исходу. Гемофилии уделялось значительное внимание в средствах массовой информации в 80-е годы XX века в связи с синдромом приобретенного иммунодефицита(СПИД). Прежде чем кровь стали тщательно проверять на наличие вируса иммунодефицита человека (ВИЧ), вызывающего СПИД, многие люди, страдающие гемофилией, при переливании крови заразились этим вирусом.
Различными способами происходит потеря половых хромосом либо образование чрезмерного их количества. Женщины могут обладать лишними Х-хромосомами, а мужчины — лишними Х- и F-хромосомами. Комбинации таких хромосом, вызывающие нарушения, приведены в табл. 3.1.
Кроме вышеперечисленных аномалий у мужчин и женщин может случаться разрыв хромосом.Примером этой врожденной генетической аномалии является синдром ослабленной Х-хромосомы.Этим термином обозначается «отламывание» небольшого участка на конце Х-хромосомы, дефект которого, хотя он исоставляет менее 1% от всей хромосомы, может привести к серьезным последствиям. Среди них такие аномалии роста, как огромная голова, большие оттопыренные уши, удлиненное лицо. Некоторых малышей, обладающих данным синдромом, отличает нетипичная манера поведения: они хлопают в ладоши, кусают свои ручки, гипе-рактивны. Сегодня ослабленная Х-хромосома является наиболее распространенным наследственным дефектом, связанным с умственной отсталостью (Tsuchiya, Forsythe, Robin & Tunnessen, 1998).
Поскольку синдром ослабленной Х-хромосомы предполагает наличие рецессивного гена в Х-хромосоме, на мужчин он обычно оказывает большее воздейст-
Глава 3. Наследственность и среда 121
вие из-за недостатка гена в У-хромосоме, который мог бы его перевесить. Тем не менее почти 20% мужчин с ослабленной Х-хромосомой не проявляют характерного для нее фенотипа. Последние исследования предполагают, что виновна в этом неустойчивая генная мутация: базовые пары рецессивного гена многократно повторяют себя, вплоть до нескольких тысяч раз. Чем больше количество повторений, тем более серьезны симптомы (Mazzocco, 2000; Sutherland, Richards, 1994; Tsuchiya et. al, 1998).
Аномалии, не зависящие от пола (аутосомные аномалии)
Аномалии, связанные с остальными 22 парами хромосом, подобно нарушениям, сцепленным с полом, могут являться результатом дефектных генов или лишних хромосом. Некоторые аутосомные нарушения обобщены в табл. 3.2.
Таблица 3.2 Примеры аномалий, не сцепленных с полом
Статистика по каждой аномалии основывается на данных о рождениях детей в США, не закончившихся смертью младенца.
ГЕНЕТИЧЕСКИЕ
СиндромЭнджелмэна
Нарушение (возможно, доминантное), имеющее место приблизительно у одного из 10-15 тысяч человек. Оно детерминируется набором мутировавших генов в 15-й хромосоме, но только в том случае, если они унаследованы от отца. В данном случае не происходит выработки нескольких протеинов, воздействующих на функционирование гипоталамуса.
Кистозный фиброз
Рецессивное нарушение, имеющее место приблизительно у одного из 2500 американцев, потомков белых европейцев; в иных случаях наблюдается реже. Среди белых американцев (США) каждый год рождается около 1500 новых детей-носителей. Мутировавший ген в 7-й хромосоме не может функционировать должным образом и разрушает несколько метаболических путей, ведущих к регуляции внешней секреции поджелудочной железы. Организм наполняется излишней слизью, в том числе — легкие и пищеварительный тракт; изменяется процесс потоотделения, так что в жаркую погоду истощаются запасы соли человека. Распространена смертность в ранней взрослости. Люди, страдающие кистозным фиброзом, должны подвергаться обширной физической терапии, чтобы освобождаться от слизи несколько раз в день, — утомительный и требующий времени процесс. Кроме того, большинство мужчин и женщин с кистозным фиброзом являются бесплодными.
Фенилкетонурия (ФКУ)
Рецессивная аномалия, имеющая место приблизительно у одного из 10 тысяч человек. Дефект гена в 12-й хромосоме не позволяет синтезировать энзим фенилалин гидрокси-лазы, ответственный за преобразование особо важного белка фенилалина в другой белок — тирозин. В отсутствие необходимого фермента фенилалин перерабатывается в фе-нилпировиноградную кислоту, которая приводит к повреждению и гибели клеток мозга. После рождения ребенка, когда энзимы матери больше не могут преобразовывать фенилалин для ребенка, эта аминокислота накапливается и мешает другим важным аминокислотам проникать в клетки. Разрушение клеток становится причиной ряда серьезных неврологических симптомов: повышенная раздражимость, неконтролируемые движения (непроизвольные подергивания мышц), гиперактивность, судорожные припадки и умственная отсталость. Сейчас в США у всех новорожденных в обязательном порядке
«I 22 Часть I. Начало
Продолжение табл. 3.2
берут анализ крови на ФКУ. Поскольку фенилалин присутствует во многих видах пищи, младенцы с ФКУ немедленно переводятся на синтетическую замену протеина, содержащую очень маленькие, но необходимые количества фенилалина. Люди с фенилкетоно-урией, проходящие такое лечение, имеют нормальные возможности для будущей жизни, могут иметь детей. Однако способные к деторождению женщины, страдающие ФКУ, подвергаются высокому риску выкидыша или нарушений родов, поскольку плод развивается в ненормальной внутриутробной среде.
Хорея Гентингтона
Доминантное нарушение, встречающееся приблизительно у одного из 10 тысяч человек. Хорея Гентингтона разносится доминантным геном в 4-й хромосоме,и поэтому может наследоваться только от одного родителя. Синтезируемый ею неверный протеин, обнаруженный в многочисленных клетках, в том числе и в нейронах головного мозга, называется гентингтон. В них он приводит к выборочной дегенерации нейронов, что, в свою очередь, вызывает деменцию, случайные дергающиеся движения, а также кособокую накрененную походку — симптомы, прогрессивно ухудшающиеся до тех пор, пока человек не становится немым, неподвижным и постепенно не умирает. Разрушение может длиться даже в течение 30 лет, хотя многие страдающие этой болезнью люди умирают гораздо быстрее от таких осложнений, как пневмония и паралич сердца (сердечная недостаточность). Это нарушение очень коварно, так как его симптомы могут не проявляться до достижения человеком 35-летнего возраста. Так, люди, постепенно растящие в себе болезнь, могут передать ее ген по наследству своим детям задолго до того, как они осознают, что являются ее носителями.
Синдром Прадера Вилли(см. текст)
Рецессивное нарушение, имеющее место приблизительно у одного из 10-15 тысяч человек. Оно детерминируется набором мутировавших генов в 15-й хромосоме, но только в том случае, если они унаследованы от матери; не вырабатываются несколько протеинов, воздействующих на функционирование гипоталамуса.
Рекомендуемые страницы:
Воспользуйтесь поиском по сайту:
Источник