Синдром срединной расщелины лица фронтоназальная дисплазия

Фронтоназальная дисплазия (ФНД) – порок развития средней части лица, заключающийся в нарушении перемещения глаз по направлению к носу в процессе эмбриогенеза. Впервые был описан в 1967 г. W. De Myer [1]. Автор изучал больных с единообразным пороком развития и назвал это заболевание синдромом срединной расщелины лица. В 1970 г. S. Sedano и соавт. предложили переименовать этот синдром в синдром фронтоназальной дисплазии, так как расщелина лица не была облигатным признаком, строго определяющим этиопатогенез синдрома [2]. Этими же авторами был описан эмбриопатогенез ФНД: порок формируется с 19-го по 21-й день эмбрионального развития из-за нарушения миграции мезодермы, обусловленного мутацией в ALX3 гене [3].
Синдром ФНД характеризуется аутосомно-доминантным типом наследования с различной пенетрантностью (проявляемостью) и экспрессивностью (степенью выраженности). При этом чаще всего встречаются спорадические случаи, как проявление мутации de novo [4].
В OMIM (Online Mendelian Inheritance in Man) имеет номер #136760. В самой масштабной работе, посвященной изучению этого синдрома, проведенной в 1996 г., рассматривался 21 случай этой редкой патологии [5]. По данным авторов, соотношение больных мужчин и женщин составляет 2:1.
Манифестные (часто встречаемые) признаки синдрома ФНД или синдромальное «ядро» заболевания (рис. 1):

Рис. 1. Патогенез синдрома ФНД [6].
Глаза: узкие глазные щели, гипертелоризм, эпи-, телекант, катаракта, дегенерация сетчатки, колобома нижнего века.
Лоб: клиновидный рост волос на лбу («мыс вдовы»), срединный дефект лобной кости (скрытая расщелина черепа, лобное менингоэнцефалоцеле).
Нос: расщелины разной степени тяжести (от раздвоенного кончика носа до полной расщелины, возможно, в сочетании с широкой срединной расщелиной верхней губы), расщелины крыльев носа, широкая переносица, отсутствие кончика носа, назальные кожные привески.
Патология центральной нервной системы: агенезия мозолистого тела.
К редким симптомам, описанным при ФНД, относят: носовые и ушные привески, микрофтальм, низко расположенные уши, кондуктивную тугоухость, липомы на лбу и в мозолистом теле. В исследовании, посвященном синдрому ФНД [6], было установлено, что агенезия мозолистого тела встречалась в 57% случаев, липома мозолистого тела – в 19% случаев. Также описаны дефект межжелудочковой перегородки, тетрада Фалло, поли-, син-, брахидактилия, расщелина позвоночника, омфалоцеле, крипторхизм [7].
Прогноз для жизни и здоровья при синдроме ФНД зависит от наличия и тяжести сопутствующих аномалий. Пороки развития лица устраняются обычно серией пластических операций. По данным литературы [6, 7], интеллект у больных ФНД обычно сохранен. Однако W. De Myer отмечает умственную отсталость у 8% и легкое снижение интеллекта у 12% больных [1].
Пренатальная диагностика синдрома фронтоназальной дисплазии
Несмотря на то что изменения фенотипа при синдроме ФНД, казалось бы, очевидны: расщелина лица, агенезия мозолистого тела, патология мягких тканей носа, гипертелоризм и др. и не должны вызывать затруднений у врача ультразвуковой пренатальной диагностики, в литературе встречается ограниченное количество публикаций, посвященных пренатальной диагностике синдрома ФНД. Редкие работы о пренатальной диагностике единичных случаев синдрома посвящены в основном применению новых технологий 3D/4D с методиками поверхностной реконструкции [8–10]. Этот факт объясняется, скорее всего, тем, что врачи выставляют диагноз отдельных симптомов данного заболевания (чаще всего это расщелина губы/неба, лобная черепномозговая грыжа, агенезия мозолистого тела, патология развития мягких тканей носа) с перечислением всех найденных при ультразвуковом исследовании пороков без попытки провести клинико-синдромальный поиск. Такой «однобокий» подход к диагностике найденных аномалий не позволяет выставить правильный клинический диагноз синдрома ФНД, что в дальнейшем приведет к неполному и неадекватному медико-генетическому консультированию (МГК) семьи, которое заключается как в определении прогноза на данную беременность, так и в формировании тактики репродуктивного поведения семьи в дальнейшем и выработке специфических мер профилактики патологии.
Медико-генетическое консультирование при синдроме ФНД
При диагностике патологии с аутосомнодоминантным типом наследования изучение фенотипа/генотипа родителей позволяет установить, явилось ли данное заболевание следствием новой мутации (de novo) либо патологический ген унаследован от кого-то из родителей.
Если у одного из родителей находят даже малейшие признаки синдрома ФНД (учитывая различную пенетрантность и экспрессивность генов, которые определяют клиническую выраженность симптомов), риск повтора данной патологии составит 50%, в случае возникновения мутации de novo этот риск не превышает уровень общепопуляционного (1%), так как члены семьи здоровы.
Клиническое наблюдение 1
При проведении пренатальной эхографии в 34 нед беременности (настоящая беременность вторая, в семье один здоровый ребенок) в медико-генетическом отделении МОНИИАГ были выявлены лицевые дизморфии у плода женского пола – гипертелоризм, раздвоенный кончик широкого носа, образование в области переносицы (лобное менингоцеле малых размеров). Выставлен пренатальный диагноз синдрома ФНД, имеющей аутосомно-доминантный тип наследования, полностью подтвержденный после родов при осмотре новорожденного генетиком-синдромологом (рис. 2).
Рис. 2. Фронтоназальная дисплазия.
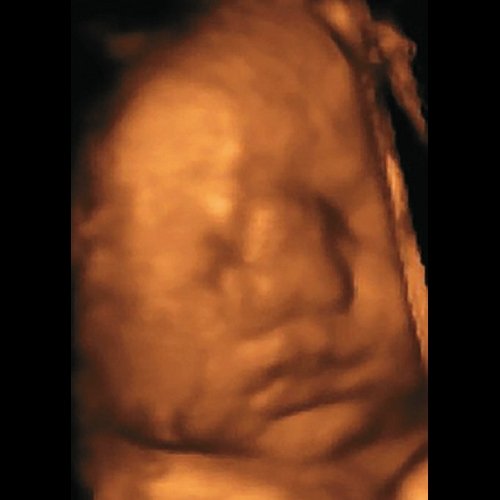
а) Пренатальный фенотип в 34 нед беременности.

б) Постнатальный фенотип новорожденного.
При составлении родословной и изучении фенотипа родителей плода выяснено, что родители здоровы и специфические проявления синдрома ФНД в изучаемых семьях не встречались. Родословная при ФНД представлена на рис. 3.
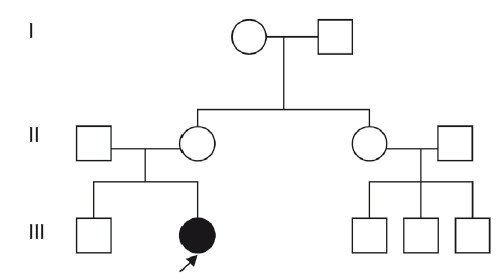
Рис. 3. Родословная при синдроме ФНД.
Исходя из этого, учитывая аутосомнодоминантный тип наследования этого синдрома, данной семье при МГК был дан благоприятный прогноз на следующие беременности, так как возникновение данного синдрома связано с мутацией de novo, при которой повторный риск рождения ребенка с этой патологией не превышает общепопуляционный. Однако родители были предупреждены, что у их девочки, являющейся носителем патологического гена, в будущем риск рождения больных детей составит 50%.
Клиническое наблюдение 2
При проведении пренатальной эхографии в 26 нед беременности (настоящая беременность первая) в медико-генетическом отделении МОНИИАГ были выявлены лицевые дизморфии у плода женского пола: гипертелоризм, раздвоенный широкий нос с образованием «гребнеобразного» выроста на его кончике. Учитывая выявленные изменения фенотипа, был выставлен пренатальный диагноз синдрома ФНД с аутосомно-доминантным типом наследования. В срок родилась девочка, у которой были подтверждены симптомы пренатально установленного генетического синдрома (рис. 4).
Рис. 4. Фронтоназальная дисплазия.
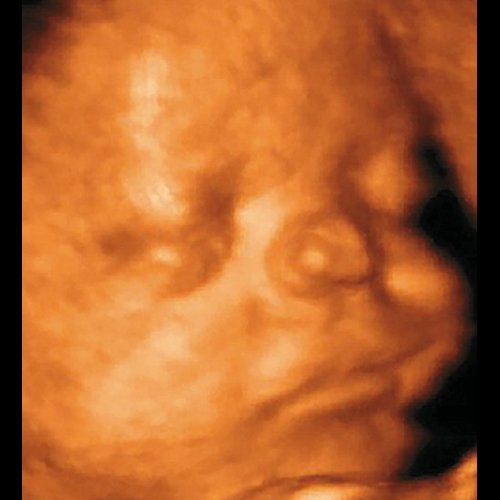
а) Пренатальный фенотип в 26 нед беременности.
б) Постнатальный фенотип новорожденного.
При изучении фенотипа родителей, несмотря на то что они считали себя здоровыми, у отца диагностированы скрытая расщелина неба, раздвоенный кончик язычка мягкого неба, большое расстояние между передними резцами (диастема). Все эти признаки являются «мягкими» признаками синдрома ФНД, т.е. отец больной девочки является носителем патологического гена, проявления которого могут быть разной степени выраженности в силу различных экспрессивности и пенетрантности. Родослов ная в данном случае синдрома ФНД представлена на рис. 5.
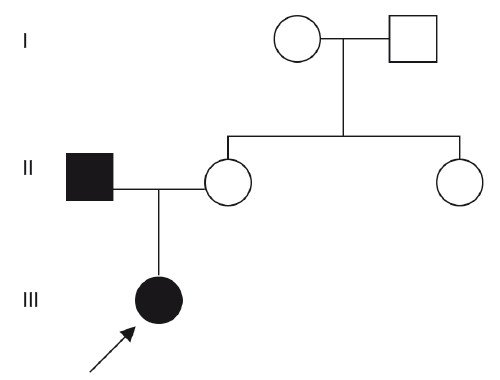
Рис. 5. Родословная при ФНД.
Исходя из вышеперечисленного, семье в данном браке при МГК был дан повторный риск 50% на все последующие беременности, так как отец является носителем патологического гена. Риск рождения больных детей у больной девочки в дальнейшем также составит 50%.
Профилактика синдрома ФНД в зависимости от происхождение заболевания
При мутации de novo риск повторного рождения ребенка с подобной патологией составляет не более 1%. В этом случае не требуется никаких специфических мероприятий при планировании следующей беременности. При выявлении генетического синдрома у кого-нибудь из родителей риск повтора заболевания у детей больного члена семьи составляет 50%. В таком случае семье рекомендуется принять решение о возможном применении вспомогательных репродуктивных технологий с донорским материалом. При наступлении самопроизвольной последующей беременности ультразвуковое исследование стоит рекомендовать проходить на экспертном уровне в медико-генетических центрах/отделениях с прицельным поиском возможных известных симптомов, встречающихся при синдроме ФНД уже со срока первого скринингового обследования (в 11–14 нед беременности).
Литература
- De Myer W. The median cleft face syndrome: differential diagnosis of cranium bifidum occultum, hypertelorism, and median cleft nose, lip and palate // Neurology. 1967; 17: 961.
- Sedano H.O., Cohen M.M., Jirasek J., Gorlin R.J. Fronto-nasal dysplasia // J Pediatr. 1970; 76: 906–913.
- Cohen M.M. Jr., Sedano H.O., Gorlin R.J., Jirasch J.E. Frontonasal dysplasia (median cleft face syndrome): comments on etiology and pathogenesis // Birth Defects Orig Artic Ser. 1971; 7: 117–119.
- Fryburg J.S., Persing J.A., Lin K.Y. Frontal dysplasia in two consecutive generations // Am J Med Genet. 1993; 46: 712–714.
- Guion-Almeida M.L., Richieri-Costa A., Saavedra D., Cohen M.M. Fronto-nasal dysplasia: analysis of 21 cases and literature review // Int J Oral Maxifac Surg. 1996; 25: 91–97.
- Кеннет Л. Джонс. Наследственные синдромы по Дэвиду Смиту. М., 2011. 283 с.
- Beryl Benacerraf. Ultrasound of Fetal Syndromes. Elsevier, 2008: 225–228.
- Shipp T.D., Mulliken J.B., Bromley B. Benacerraf B. Three-dimensional prenatal diagnosis of frontonasal malformation and unilateral cleft lip/palate // Ultrasound Obstet Gynecol. 2002; 20: 290–293.
- Schoonveld C., Yamamura Y., All M., Veres J., Ramin K.D. 3D ultrasound enhances congruence of prenatal and postnatal diagnosis of frontonasal dysplasia // Ultrasound Obstet Gynecol. 2008; 32: 461.
- Johnson J.M., Benoit B., Pierre-Louis J., Keating S., Chitayat D. Early prenatal diagnosis of oculoauriculofrontonasal syndrome by threedimensional ultrasound // Ultrasound Obstet Gynecol. 2005; 25: 184–186.
При поддержке гранта РГНФ № 15-06-10977/15
УЗИ аппарат HS40
Лидер продаж в высоком классе. Монитор 21,5″ высокой четкости, расширенный кардио пакет (Strain+, Stress Echo), экспертные возможности для 3D УЗИ в акушерско-гинекологической практике (STIC, Crystal Vue, 5D Follicle), датчики высокой плотности.
Источник
Атипичные лицевые расщелины встречаются значительно реже по сравнению с расщелинами губы и нёба. Иногда расщелины губы и нёба сочетаются с различными формами редких лицевых расщелин, такими как колобома века, поперечными и косыми расщелинами лица или бывают одним из симптомов врожденного синдро¬ма (синдром Ван-дер-Вуда, Пьера Робена и др.). В 15-20% случаев они являются компонентами наследственных синдромов. В клинической практике ряд врожденных деформаций черепно-лицевой области и синдромов сопровождаются нетипичными (редкими) расщелинами. Среди них выделяются: Поперечная расщелина лица (макростома, 7,8 по Tessier) бывает одно- и двусторонней. Возникает как результат несращения верхнечелюстного и нижнечелюстного бугров в период эмбрионального развития. Клинически патология проявляется в виде макростомы различной степени выраженности, при этом расщелина начинается от угла рта и продолжается далее по направлению к мочке уха. Макростома может быть как изолированным пороком развития, так и симптомом некоторых врожденных синдромов. К синдромам бранхиальных дуг относятся синдромы: гемифациальной микросомии, синдром Тричера Коллинза — Франческетти, синдром Нагера, синдром Мюллера. Поражения челюстно-лицевой области характерные для синдрома Коллинза-Франческетти отмечаются у пациентов с акрофациальным синдромом Нагера и синдромом Миллера. Отличительным признаком синдрома Нагера является сопутствующая патология верхних конечностей (гипо- или аплазия I пальца и лучевой кости, нарушение функции локтевого сустава). Синдром Миллера является очень редким генетическим заболеванием. Характеризуется более тяжелыми повреждениями конечностей: гипоплазия или отсутствие костей предплечья или голени, пальцев на руках и ногах, синдактилия. |
Источник