Синдром сэтре чотзена что это

Краткие сведения о нозологии
Выделяют следующие синдромы: синдром Апера, синдром Крузона, синдром Пфайффера, синдром Сэтре–Чотзена, синдром Джексона–Вейса, синдром Карпентера и др.
Клинические синдромы:
Синдром Апера (Apert syndrome)
Заболевание характеризуется КС коронарных швов с формированием брахицефалии или акроцефалии, вдавленной деформацией средней зоны лица и симметричной синдактилией конечностей. Частота возникновения СА составляет от 7,6 до 22 случаев на 1 млн живорожденных, причем у азиатов частота встречаемости самая высокая, а у испанцев – самая низкая. Наследование СА происходит по аутосомно- доминантному типу и вызывается мутацией гена рецептора фактора роста фибробластов 2-го типа (fibroblast growth factor receptor 2 – FGFR2), расположенного на длинном плече хромосомы 10. Одной из отличительных особенностей пациентов с СА является макрокрания, которая сочетается с КС.
Другой особенностью СА является преждевременное смыкание сфеноокципитального и петроокципитальных синхондрозов.
Нередко у пациентов обнаруживают пороки развития головного мозга, такие как дистопия миндалин мозжечка, стеноз яремного отверстия, арахноидальные кисты в задней черепной ямке, мальформации мозолистого тела и/или лимбических структур.
Следует заметить, что прогрессирующая гидроцефалия неспецифична для заболевания, за нее ошибочно принимают непрогрессирующую вентрикуломегалию, часто наблюдаемую у этих больных. У пациентов с СА обычно наблюдается умственное недоразвитие разной степени выраженности, однако имеются пациенты и с нормальным интеллектом. К основным лицевым признакам СА относится окулярный проптоз (экзорбитизм), который может быть асимметричным. Поражения органа зрения. Гипоплазия верхней челюсти,расщелина язычка или мягкого нёба, расщелина твердого нёба, альвеолярного отростка верхней челюсти и губы очень редки. Задержка прорезывания зубов. Кроме описанных пороков развития, примерно у 2/3 пациентов наблюдается срастание шейных позвонков. Также описаны пороки развития трахеи.
Синдром Крузона (Crouzon syndrome)
Синдром Крузона (СК) является типичным КС с вовлечением не только коронарного, но и сагиттального и лямбдовидного швов. Порок сопровождается выраженной гипоплазией средней трети лица, с очевидным окулярным проптозом, но при этом заболевании не выявляют грубых пороков развития кистей и стоп, что отличает его отдругих заболеваний этой группы. СК является самым частым в группе синдромальных КС и наблюдается у 1 из 65 000 новорожденных. Так же как и СА он наследуется по аутосомно-доминантному типу и вызывается мутацией гена FGFR2. Обычно к моменту рождения уже синостозировано несколько швов черепа, а в течение первых лет жизни количество включенных в процесс швов может увеличиться. Преждевременное смыкание сфеноокципитального и петрозоокципитального синхондрозов при СК наблюдается часто и происходит в конце внутриутробного периода или вскоре после рождения. Форма головы зависит от того, какие швы и в какой последовательности синостозировались. Кости черепа обычно тонкие, с отчетливыми пальцевыми вдавлениями. Передняя, средняя и задняя черепные ямки короткие, но при этом в отличие от СА практически всегда симметричные. Пороки развития ЦНС наблюдаются у пациентов с СК реже, чем при СА. Они включают аномалию Киари 1-го типа и прогрессирующую гидроцефалию. Стеноз яремных отверстий в сочетании с сужением яремных вен наблюдается в 60% случаев. Более выраженный симметричный экзорбитизм. Верхняя челюсть сильно недоразвита, что в сочетании с костным дефицитом нижнего края орбит усиливает окулярный проптоз. Нёбо узкое, высокое, у половины больных отмечаются латеральные утолщения слизистой оболочки. Нарушения окклюзии и скученность зубов на фоне гипоплазии верхней челюсти так же являются характерными признаками порока, однако в отличие от СА значительной задержки прорезывания зубов нет. Сращение шейных позвонков у 22% больных. Примерно у половины — кондуктивная тугоухость. У 13% имеется стеноз или атрезия наружного слухового канала. Часто наблюдаются значительные нарушения дыхания.
Синдром Пфайффера (Pfeiffer syndrome)
Типичными проявлениями синдрома Пфайффера (СП) являются коронарный КС, гипоплазия
верхней челюсти, экзорбитизм и пороки развития конечностей, такие как широкие большие пальцы
рук и ног, синдактилия и брахидактилия. При СП выявляются мутации генов FGFR1
или FGFR2. Наследуется синдром по аутосомнодоминантному типу. Частота синдрома не
определена из-за его редкости. Выделяют три типа синдрома.
Тип I описывается как классическая или мягкая форма заболевания. Он характеризуется КС коронарного и нередко сагиттального шва, с формированием брахицефалии. Деформация черепа и лица при этом типе напоминает таковую при мягком фенотипе СА. Пациенты с этим типом обычно имеют нормальный или близкий к норме уровень интеллекта и обычную продолжительность жизни.
Тип II более тяжелый и характеризуется КС многих швов. В основном зарастают коронарный, лямбдовидный и метопический швы с компенсаторным ростом черепа по линии открытых сагиттального и височно-теменных швов, что придает голове форму трилистника (череп в виде листа клевера – Cloverleaf skull). При этом типе может отмечаться тяжелый экзорбитизм. Интеллект у детей часто снижен, одним из характерных признаков является плечелучевой синостоз. При отсутствии лечения продолжительность жизни детей с этим типом СП невелика.
Тип III по тяжести черепно-лицевых проявлений похож на тип II, за исключением того, что КС не приводит к формированию трехдольчатого черепа. Большинство пациентов фенотипически напоминают СК, от которого их отличают характерные деформации больших пальцев руки ног. Также у них наблюдается плечелучевой синостоз. Характерными для всех типов СП являются типичные деформации больших пальцев рук и ног в виде их укорочения, утолщения и клинодактилии. Другими признаками являются кожная синдактилия, чаще пальцев стоп и брахидактилия кистей и стоп за счет укорочения средних фаланг. Все пациенты с СП имеют гипоплазию средней зоны лица в сочетании со скученностью зубов и нарушениями прикуса.
Синдром Джексона–Вейса (Jackson–Weiss syndrome)
Синдром Джексона–Вейса характеризуется вовлечением в патологический процесс нескольких швов черепа с формированием акроцефалии. Гипоплазия средней трети лица также типична для этого заболевания, но выражена не так сильно, как при СК. Еще одним характерным признаком являются утолщение и девиация больших пальцев стоп, как при СП, большие пальцы рук при этом не поражаются. Наследуется заболевание по аутосомно-доминантному типу с вариабельной экспрессивностью. Мутация происходит в гене FGFR2. Интеллектуальное развитие часто не страдает. Хотя и описаны индивидуумы с низким или пограничным уровнем интеллекта.
Синдром Карпентера (Carpenter syndrome)
Типичными признаками синдрома Карпентера являются КС и полисиндактилия стоп. В отличие от СА, при котором также описана постаксиальная полидактилия (полидактилия мизинца), при синдроме Карпентера полидактилия всегда преаксиальная. Череп при этом заболевании деформирован по типу акроцефалии и не имеет характерных отличий от других синдромальных КС. Заболевание наследуется по аутосомно-рецессивному типу и его связывают с дефектом RAB23 из группы Ras-генов, который является негативным регулятором сигнальной системы группы НН (hedgehog). Другими признаками болезни являются низкий рост, ожирение, врожденные пороки сердца, задержка интеллектуального развития. Выбор тактики и сроков лечения напрямую зависит от тяжести внутричерепной гипертензии.
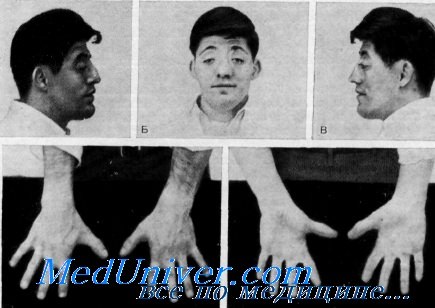
Синдром Сэтре–Чотзена (Saethre–Chotzen syndrome)
Синдром Сэтре–Чотзена (ССЧ) единственный из описанных выше синостозов характеризуется асимметрией черепа и лица. Основные проявления ССЧ включают КС, птоз верхних век, высокий лоб с низкой линией роста волос и пороки развития конечностей, такие как брахидактилия и мягкотканная синдактилия. ССЧ наследуется по аутосомно-доминантному типу с высокой пенетрантностью и обусловлен главным образом мутацией гена TWIST. КС наблюдаются у большинства, но не у всех пациентов, чаще всего срастается коронарный шов, приводя к брахицефалии или акроцефалии. Также отмечено зарастание лямбдовидного и метопического швов. Для ССЧ характерны позднее закрытие родничков, увеличенные отверстия теменных выпускников и другие дефекты костей черепа. Длина передней черепной ямки укорочена, а турецкое седло может быть углублено. Из лицевых признаков ССЧ отмечают широкое уплощенное основание носа с искривлением носовой перегородки, нос может быть длинным, тонким, с заостренным или крючковидным кончиком. Верхняя челюсть у таких больных часто недоразвита, что вместе с высоким лбом формирует уплощенный лицевой профиль. Нёбо часто сужено, с высоким сводом и иногда с расщелиной. Аномалии зубов включают сверхкомплектные зубы, гипоплазию эмали и нарушения образования дентина, приводящие к истончению и сужению корней. Деформации конечностей включают частичную мягкотканную синдактилию между II и III пальцами кисти или стопы. Также наблюдаются брахидактилия, клинодактилия и раздвоение дистальных фаланг.
Диагностика:
МСКТ костей черепа и головного мозга с трехмерной реконструкцией наиболее информативный метод диагностики. Позволяет достоверно выявить преждевременно заросший шов, подтвердить, клинически выявленную, деформацию формы головы.
УЗИ костей черепа так же позволяет выявить преждевременное заращение швов черепа.
Показания к оперативному лечению:
- Краниосиностоз
- Повышение внутричерепного давления
- Деформация челюстно-лицевой системы
Противопоказания к оперативному лечению:
- Воспалительный процесс любой локализации в стадии обострения, неполной ремиссии
- Анемия средней и тяжелой степени.
- Тяжелое общесоматическое состояние
- Обострение хронических заболеваний.
Окончательное решение о наличии или отсутствии показаний к оперативному лечению принимает врач-нейрохирург в рамках очной консультации.
Примерное количество койко-дней: 8-10 койко-дней.
Описание результата операции:
Особенности послеоперационного периода:
Ограничения в послеоперационном периоде/
Необходимость в повторной явке в ФЦН. В послеоперационном периоде повторная консультация нейрохирурга не реже 1 раз в 6 мес.
Источник
Глазо-зубо-костная дисплазия. Синдром Пфейффера и Сетре—ЧотценаГлазо-зубо-костная дисплазия — синдром, включающий узкий нос с гипопластичными крыльями, микрокорнеа с аномалией радужной оболочки, синдактилию мягких тканей IV и V пальцев, недостаточность формирования длинных костей и гипоплазию эмали. Среди 45 случаев было 3 больных с проводящей глухотой различной степени (Gorlin et al., Gillespie, Reisner et al.). Синдром Сетре—Чотцена (Saethre—Chotzen) и проводящая глухота. Синдром Сетре — Чотцена характеризуется краииосиностозом, низкой линией роста волос на лбу, асимметрией лица, птозом одного или обоих век, смещением носовой перегородки и разными кожными синдактилиями. Согласно данным Pantkc с соавт., к настоящему времени опубликовано около 85 случаев синдрома. Авторы лично обследовали 31 больного. Bartsocas с соавт. представили хороший литературный обзор.
Pantke сообщил, что у 15% больных имеется легкая проводящая глухота, в некоторых случаях односторонняя. Далее он отметил, что дефект слуха был выявлен у 11 из 22 больных, которым было произведено аудиометрическое обследование. В большинстве этих случаев тип и/или степень глухоты не указаны. Однако Chotzen и Hammar с Roggenkamp отметили проводящую глухоту умеренной степени. Grebe обнаружил глухонемоту, но тип потери слуха не определил. Dolivo и Gillieron сообщили о смешанной глухоте. В наблюдении Jorgenson диагноз является менее определенным, но у ребенка наблюдалась проводящая глухота. Синдром фокальной гипоплазии кожи и смешанной глухоты. Фокальная гипоплазия кожи характеризуется атрофией и линейной гиперпигмептацией поверхностного слоя кожи, ограниченным отложением в коже жира, множественными папилломами слизистых оболочек или кожи вокруг рта, атрофией ногтей и многочисленными скелетными аномалиями. Это заболевание описано Goltz с сотр. и Ginsburg с соавт.. Наследование, вероятно, Х-сцепленное доминантное, летальное для мальчиков. По-видимому, от 5 до 10% больных с этим синдромом имеют дефект слуха. Исследования височной кости не проводились. Holden и Akers сообщили о смешанной глухоте, выявленной у их больного в возрасте 3 лет. St oilman кратко отметил наличие нейросенсорной глухоты у своего больного. Goltz с соавт. также обнаружили смешанную глухоту. Daly, Ginsburg с соавт. и Ferrara описали сужение наружного слухового прохода. Глазо-глоточная мышечная дистрофия и нейросенсорная глухота. Это одно из нескольких заболеваний, при которых наблюдается миопатия лица. Выявляется оно поздно, обычно в середине жизни. Слабость лицевой мускулатуры достигает степени вялого паралича. Вовлечение в процесс жевательных мышц ведет к западению височных областей и отвисанию нижней челюсти. Резко выражен птоз. Лоб обычно испещрен бороздами. Нарастают затруднения в приеме пищи и жидкости. Заболевание характеризуется аутосомyо-доминантным наследованием. В одной семье обнаружена медленно прогрессирующая нейросенсорная глухота (Graf). Conversely, Roberts и Bamforth не отметили глухоты среди 26 больных с этим заболеванием. Kuhn и Еу обнаружили глухоту у 8 из 23 больных с миотонической дистрофией, у 4 из них нарушения слуха возникли в связи с инфекцией среднего уха. — Также рекомендуем «Синдром Варденбурга: клиника, диагностика» Оглавление темы «Наследственные болезни с глухотой»:
|
Источник
Двухлетнего мальчика спасет реконструкция черепа
Никита Лапшин, 2 года, синдром Сетре – Чотзена (деформация черепа, асимметрия лица), спасет операция, требуется подготовка к ней и саморассасывающиеся пластины. Собрано 866 413 руб. Госпитализация в январе–2020.
Как помочь
До рождения сына ничто не предвещало беды – беременность у меня прошла нормально, я хорошо себя чувствовала, все анализы и тесты были в норме. А когда сын родился, меня поразила форма его головы: она была вытянута вверх башенкой, лоб большой, выпуклый, а затылок плоский. И ушные раковины неправильно сформированы. Личико тоже выглядело странно, черты асимметричные. Из роддома нас перевели в детскую больницу, начали обследовать. Выявили множество врожденных пороков: аномалию развития сердца, микрофтальм (уменьшение глазного яблока) и даже глубокую поперечную борозду на ладони, врачи говорили, что это признак генетического синдрома. Даже синдром Дауна у сына заподозрили, но, к счастью, генетический анализ этот диагноз не подтвердил. Невролог направил нас на консультацию к нейрохирургу, который выявил у Никиты двусторонний коронарный синостоз – брахицефалию. Это означало, что черепные кости срослись еще внутриутробно, это и стало причиной деформации черепа. И кости лица также сформировались неправильно: у Никиты обнаружили нарушение строения верхней челюсти и носовой перегородки. На основании этих признаков врачи предположили редкую аномалию: синдром Сетре – Чотзена.

Мы повезли Никиту в Москву, здесь его обследовали и диагноз подтвердили. Тогда сыну срочно требовалась операция для устранения деформации черепа. Подготовку к ней и специальные аппараты нужно было оплатить, у нас такой возможности не было. К счастью, на помощь пришел Русфонд. Мы очень благодарны всем, кто откликнулся! Операция прошла успешно, но остается еще дефект лобной кости. Его нужно устранить и закрыть специальными биопластинами, операцию также вначале смоделируют на компьютере. Все это стоит очень дорого, и мы по-прежнему нуждаемся в вашей помощи. У нас двое детей, живем на скромную зарплату мужа – он прапорщик, служит у нас в Нижнем.
Никита у нас растет активным и любознательным, он пока разговаривает на своем языке, но все-все понимает, любит смотреть мультики и танцевать под ритмичную музыку. А еще сынок очень хозяйственный, старается мне помогать, научился запускать стиральную машину и всегда убирает за собой посуду.
Пожалуйста, не оставляйте нас, помогите вылечить Никиту!
Ольга Лапшина,
Нижегородская область
Фото из архива семьи
Подготовила Мария Стояновская

Руководитель Центра челюстно-лицевой хирургии Виталий Рогинский (Москва): «Мальчик был успешно прооперирован год назад, череп приобрел более округлую форму. Но сохраняется дефект лобной кости. Его нужно закрыть с помощью биорезорбируемых (саморассасывающихся) пластин. Госквота выделена только на операцию, подготовка к ней и пластины квотой не покрываются».
Стоимость подготовки к операции и саморассасывающихся пластин 690 000 руб. 60 000 руб. внес один московский фонд, пожелавший остаться неназванным. 806 413 руб. собрали наши читатели.
Дорогие друзья! Если вы решили спасти Никиту Лапшина, пусть вас не смущает цена спасения. Любое ваше пожертвование будет с благодарностью принято. Можно воспользоваться и нашей системой электронных платежей, сделав пожертвование с банковской карты или электронной наличностью, в том числе и из-за рубежа. А владельцы айфонов и телефонов на платформе андроид могут отправить пожертвование через мобильное приложение. Скачать его можно здесь.
Экспертная группа Русфонда
Источник