Синдром сильвера рассела код по мкб

Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Описание
- Причины
- Симптомы
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Синдром Рассела-Сильвера.
Синдром Рассела-Сильвера
Описание
Синдром Рассела-Сильвера врожденное заболевание, характеризующееся внутриутробной задержкой роста плода, поздним закрытием большого родничка, нарушением формирования скелета. Тип наследования неизвестен. Встречается в большинстве случаев спорадически, однако описаны и единичные родословные с данной патологией. Частота в популяции 1:30 000. Встречается в равной степени, как у мужчин, так и у женщин.
Причины
Причиной заболевания считаются генетические нарушения, приводящие к подобным порокам развития. Дети с данной патологией имеют малую длину и массу тела при рождении. Значительная задержка роста и костного созревания наблюдается и в дальнейшем.
Главной причиной преждевременного полового созревания при этом заболевании является избыток гонадотропных гормонов.
Симптомы
Дети рождаются небольшой длины (до 45 см) и с малой массой тела (1,5—2,5 кг). С годами отставание в росте сохраняется, в связи, с чем окончательный рост у женщин менее 150 см, у мужчин немногим выше 150 Масса тела у взрослых нормальная или даже избыточная. Часты аномалии мочевыделительной системы и наружных половых органов: крипторхизм, гипоспадия, гипоплазия полового члена, мошонки. Характерна асимметрия тела (лицо, туловище, длина ног). Лицо треугольной формы: псевдогидроцефалия, большой лоб и гипоплазия нижней челюсти, высокое нёбо, нередко с расщелиной, оттопыренные уши. Узкая грудная клетка, короткие руки, поясничный лордоз. Половое развитие опережает возраст ( в 30% случаев), начинается в 5-6 лет и обусловлено гонадотропной стимуляцией яичников. Интеллект сохранен в большинстве случаев.
Лечение
С 1985 г. Для лечения детей с соматотропной недостаточностью используются исключительно генно-инженерные препараты гормона роста человека.
В настоящее время в России прошли клиническую апробацию и разрешены к использованию следующие рекомбинантные препараты гормона роста человека: Нордитропин RНордиЛетR (Ново Нордиск, Дания); хуматроп (Лилли Франс, Франция); генотропин (Пфайзер Хелс АБ, Швеция); сайзен (Индустрия Фармасьютика Серано С. А. , Италия); растан (Фармстандарт, Россия).
При лечении гипофизарного нанизма у детей имеется четкая связь «доза–ростовой эффект», особенно выраженная в первый год лечения. Рекомендуемая стандартная доза СТГ при терапии классического дефицита СТГ – 0, 033 мг/кг/на инъекцию, ежедневно, подкожно, в вечернее время 20,00-22,00.
Критерием эффективности терапии является увеличение скорости роста от исходной в несколько раз. Она достигает в первый год лечения, по данным разных авторов, от 8 до 13 см в год. Максимальная скорость роста отмечается в первый год лечения, особенно в первые 3-6 мес, затем имеет место замедление скорости роста от первого ко второму году лечения (при сохранении скорости роста более 5-6 см в год).
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Синдро́м Рассе́ла-Си́львера (карликовость Рассела-Сильвера) — комплекс наследственных аномалий (предполагается аутосомно-доминантный тип наследования[2]) врождённый карликовый рост в результате нарушений эмбрионального развития: малая масса тела при рождении, низкий рост, задержка общего развития, треугольное лицо, опущенные вниз уголки рта, укороченные и согнутые пальцы рук, синдактилия[3].
История[править | править код]
Впервые заболевание описано в середине прошлого века.
Эпидемиология[править | править код]
Частота в популяции 1:300 000. Встречается в большинстве случаев спорадически, однако описаны и единичные родословные с данной патологией. Является не наследственной патологией, хотя известны единичные случаи. Передается с дефектными генами.
Клиническая картина[править | править код]
Основные проявления: пренатальная (внутриутробная) задержка роста, асимметрия тела, значительное отставание массы тела от нормы, клинодактилия V пальца, псевдогидроцефалия, треугольное лицо, крипторхизм[4], гипогликемия, повышенный уровень ЛГ и ФСГ[5].
Дети рождаются небольшой длины (до 45 см) и с малой массой тела (1,5—2,5 кг). С годами отставание в росте сохраняется, в связи с чем окончательный рост у женщин менее 150 см, у мужчин немногим выше 150 см. Масса тела у взрослых нормальная или даже избыточная. Часты аномалии наружных половых органов: крипторхизм, гипоспадия, гипоплазия полового члена, мошонки. Характерна асимметрия тела (лицо, туловище, длина ног). Лицо треугольной формы: псевдогидроцефалия, большой лоб и гипоплазия нижней челюсти, высокое нёбо, нередко с расщелиной, оттопыренные уши. Клинодактилия V пальцев за счёт девиации дистальной фаланги, узкая грудная клетка, короткие руки, поясничный лордоз[4].
Часто аномалии мочевыделительной системы. В литературе описаны случаи сочетания карциномы печени и данного синдрома. Интеллект обычно нормальный[4].
Диагностика[править | править код]
Основывается на характерной клинической симптоматике[4].
Лечение[править | править код]
Симптоматическое[4].
Примечания[править | править код]
- ↑ Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- ↑ SILVER-RUSSELL SYNDROME. OMIM.
- ↑ Симптомы и синдромы в эндокринологии / Под ред. Ю. И. Караченцева. — 1-е изд. — Х.: ООО «С.А.М.», Харьков, 2006. — С. 137-138, 150. — 227 с. — (Справочное пособие). — 1000 экз. — ISBN 978-966-8591-14-3.
- ↑ 1 2 3 4 5 Малая энциклопедия врача-эндокринолога / Под ред. А. С. Ефимова. — 1-е изд. — К.: Медкнига, ДСГ Лтд, Киев, 2007. — С. 335-336. — 360 с. — («Библиотечка практикующего врача»). — 5000 экз. — ISBN 966-7013-23-5.
- ↑ Эндокринология / Под ред. Н. Лавина. — 2-е изд. Пер. с англ. — М.: Практика, 1999. — С. 72, 140. — 1128 с. — 10 000 экз. — ISBN 5-89816-018-3.
Источник
Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Синонимы диагноза
- Описание
- Причины
- Патогенез
- Симптомы
- Лечение
Другие названия и синонимы
Синдром гипотонии-ожирения.
Названия
Синдром Прадера — Вилли.
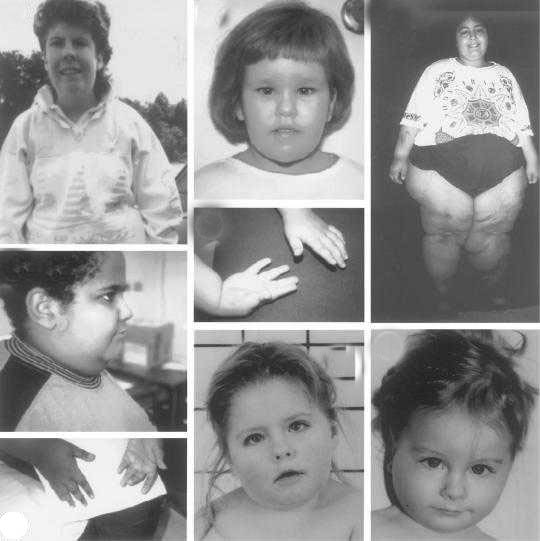
Проявления синдрома Прадера — Вилли
Синонимы диагноза
Синдром гипотонии-ожирения.
Описание
Синдром Прадера — Вилли — редкая генетическая аномалия. При синдроме Прадера — Вилли отсутствуют или не экспрессируются примерно 7 генов из 15-й хромосомы, унаследованной от отца.
Кариотип 46 XX или ХУ, 15q-11-13. Заболевание впервые описано швейцарскими педиатрами А. Prader и H. Willi в 1956 г.
По данным регистра ассоциации больных с синдромом Прадера-Вилли, в США и Канаде на декабрь 1986 г. Насчитывалось 1595 больных. В последние годы удалось установить популяционную частоту патологии, составляющую 1 : 10 000 — 1 : 20 000.
Причины
Авторы, впервые описавшие синдром, высказывали предположение об аутосомно-рецессивном типе наследования заболевания. Затем появились сообщения о возможности аутосомно-доминантной передачи болезни. Подтверждением данных гипотез могли служить наблюдавшиеся семейные случаи патологии. Однако большинство описанных клинических наблюдений синдрома Прадера — Вилли носило спорадический характер.
Последующие исследования позволили установить у детей с синдромом Прадера — Вилли определенные хромосомные нарушения. Цитогенетический анализ показал, что хромосомные аномалии у больных были представлены либо транслокациями (t 15/15), либо мозаицизмом. В 1987 г. Появились первые сообщения о микроделеции хромосомы 15. Однако окончательная идентификация хромосомных изменений при синдроме Прадера — Вилли стала возможной только после внедрения в практику молекулярно-генетических методов исследования.
В настоящее время установлено, что развитие синдрома Прадера — Вилли связано с повреждением критического района хромосомы 15 (сегмента q11,2- q13). При этом оказалось, что повреждение этого же участка хромосомы 15 наблюдается и при другом заболевании — синдроме Ангельмана, клиническая картина которого существенно отличается от синдрома Прадера — Вилли и характеризуется ранним (в возрасте 6-12 мес) замедлением психомоторного развития, микроцефалией, нарушением речи (в 100% случаев), атаксией, неконтролируемым насильственным смехом, частыми эпилептиформными припадками, специфическим выражением лица.
Таким образом, несмотря на повреждение при синдромах Прадера — Вилли и Ангельмана одного и того же локуса хромосомы 15, клинические проявления обеих болезней резко противоположны.
Объяснение фенотипических различий получено лишь в последние годы. Оказалось, что развитие этих заболеваний связано с новыми генетическими явлениями — геномным импринтингом и унипарентальной дисомией.
Геномный импринтинг — новое явление, открытое благодаря успехам молекулярной генетики. Он означает различную экспрессию генетического материала (гомологичных аллелей) в хромосомах в зависимости от отцовского или материнского происхождения, т. Е. Свидетельствует о влиянии родителей на фенотип ребенка. До настоящего времени считалось, что вклад в проявляемость (экспрессию) генов отца и матери равноценен.
По сути геномный импринтинг — это половой и тканевозависимый сложный модификатор генной активности некоторых локусов хромосом в зависимости от их родительского происхождения. Проявления геномного импринтинга выявлены и при других заболеваниях — синдромах Сотоса, Беквита-Видемана, Сильвера-Рассела, муковисцидозе и других.
Унипарентальная (однородительская) дисомия — наследование обеих хромосом только от одного из родителей. В течение многих лет считалось, что такое наследование невозможно. Лишь с помощью молекулярно-генетических маркеров удалось доказать возможность однородительской дисомии. Природа унипарентальной дисомии окончательно не выяснена, однако установлено, что она обязана своим происхождением ряду генетических и биохимических нарушений.
Следует отметить, что с помощью обычного исследования хромосомного состава кариотипа выявить микроделецию или унипарентальную дисомию невозможно. Для этого применяются специальные цитогенетические и молекулярно-генетические методы — прометафазный анализ, использование ДНК-маркеров определенных участков хромосомы 15 (исследование процессов метилирования) и.
На сегодняшний день синдромы Прадера — Вилли и Ангельмана служат общепринятой моделью для изучения новых в клинической генетике и сложных явлений — геномного импринтинга и унипарентальной дисомии.
Установлено, что синдром Прадера — Вилли может быть обусловлен двумя основными механизмами. Первый из них — микроделеция хромосомы 15 (15q11,2-q13), которая всегда отцовского происхождения. Второй — материнская изодисомия, т. Е. Когда обе хромосомы 15 получены от матери. Развитие синдрома Ангельмана, наоборот, связано с микроделецией того же участка хромосомы 15, но материнского происхождения, или отцовской изодисомией. Большинство (около 70%) случаев синдрома Прадера — Вилли обусловлено микроделецией, остальные — дисомией. При этом обращает на себя внимание отсутствие клинических различий между больными с микроделецией и изодисомией.
Патогенез
Патогенез синдрома Прадера — Вилли до настоящего времени остается малоисследованным. Высказываются предположения, что ожирение у больных обусловлено значительным (более чем в 10 раз) усилением синтеза жира из ацетата и крайне низкими процессами липолиза.
Гипогонадизм по гипогонадотропному типу может быть связан с дисфункцией гипоталамуса, преимущественно, в области вентромедиального и вентролатерального ядер. Правильность данной точки зрения подтверждается эффективностью лечения больных фармацевтическими препаратами (кломифен), приводившими к увеличению в плазме содержания лютеинизирующего гормона, тестостерона, нормализации показателей почечной экскреции гонадотропинов, сперматогенеза и появлению вторичных половых признаков.
Одним из объяснений гипопигментации кожи, волос и радужки служит снижение активности тирозиназы в волосяных фолликулах и меланоцитах, а также уменьшение пигмента в сетчатке.
Обращается внимание на повышенный риск развития лейкемии у больных с синдромом Прадера — Вилли. Исследования выявили снижение репарации ДНК (до 65% по сравнению с 97% у здорового ребенка) в лимфоцитах больных с данной патологией. Не исключено, что низкая репарационная способность ДНК может играть роковую роль в развитии злокачественных новообразований у лиц с синдромом Прадера — Вилли.
Симптомы
Дети с синдромом Прадера — Вилли обычно рождаются доношенными с незначительной внутриутробной гипотрофией и нередко в асфиксии. В 10-40% случаев наблюдается ягодичное предлежание.
В течение заболевания можно выделить две фазы: первая — свойственна детям 12-18 мес жизни. Она характеризуется выраженной мышечной гипотонией, снижением рефлексов — Моро, сосательного и глотательного, что затрудняет кормление ребенка. Вторая — наступает позже, через несколько недель или месяцев. Появляются полифагия, постоянное чувство голода, приводящие к развитию ожирения, причем отложение жира наблюдается преимущественно на туловище и в проксимальных отделах конечностей.
Мышечная гипотония постепенно уменьшается и к школьному возрасту почти полностью исчезает. Стопы и кисти больных диспропорционально маленькие — акромикрия. У детей отмечается гипогонадизм (у мальчиков — гипоплазия полового члена, мошонки, крипторхизм, а у девочек — недоразвитие половых губ и в 50% случаев — матки).
Рост больных нередко снижен. У 75% детей наблюдается гипопигментация кожи, волос и радужки. Часто диагностируется микроцефалия. Психомоторное развитие отстает от возрастной нормы — коэффициент интеллектуального развития — от 20 до 80 ед. (при норме 85-115 ед. ). Речь затруднена, словарный запас уменьшен. Больные доброжелательны, настроение характеризуется частой сменой. Описаны нарушения координации, судороги, страбизм.
Встречаются и другие аномалии: микродонтия, гипоплазия хрящей ушных раковин, сколиоз, эктропион (выворот века), глаукома.
Нередко развитие сахарного диабета, который с возрастом имеет тенденцию к улучшению.
При морфологическом исследовании мозга и ЯМР-томографии могут наблюдаться (примерно в 12% случаев) кисты червя мозжечка, аномалии коры головного мозга.
Продолжительность жизни больных может достигать 60 лет и более.
Лечение
Терапия синдрома Прадера — Вилли окончательно не разработана. По данным литературы, комплекс лечебных мероприятий включает лишь диету с ограничением жиров и углеводов и препараты, способствующие формированию вторичных половых признаков (гонадотропины).
Источник
Синдро́м Рассе́ла-Си́львера (карликовость Рассела-Сильвера) — комплекс наследственных аномалий (предполагается аутосомно-рецессивный тип наследования) врождённый карликовый рост в результате нарушений эмбрионального развития: малая масса тела при рождении, низкий рост, задержка общего развития, треугольное лицо, опущенные вниз уголки рта, укороченные и согнутые пальцы рук, синдактилия[1].
История
Впервые заболевание описано в середине прошлого века.
Эпидемиология
Частота в популяции 1:30 000. Встречается в большинстве случаев спорадически, однако описаны и единичные родословные с данной патологией. Тип наследования не установлен[2].
Клиническая картина
Основные проявления: пренатальная (внутриутробная) задержка роста, асимметрия тела, значительное отставание массы тела от нормы, клинодактилия V пальца, псевдогидроцефалия, треугольное лицо, крипторхизм[2], гипогликемия, повышенный уровень ЛГ и ФСГ[3].
Дети рождаются небольшой длины (до 45 см) и с малой массой тела (1,5—2,5 кг). С годами отставание в росте сохраняется, в связи с чем окончательный рост у женщин менее 150 см, у мужчин немногим выше 150 см. Масса тела у взрослых нормальная или даже избыточная. Часты аномалии наружных половых органов: крипторхизм, гипоспадия, гипоплазия полового члена, мошонки. Характерна асимметрия тела (лицо, туловище, длина ног). Лицо треугольной формы: псевдогидроцефалия, большой лоб и гипоплазия нижней челюсти, высокое нёбо, нередко с расщелиной, оттопыренные уши. Клинодактилия V пальцев за счёт девиации дистальной фаланги, узкая грудная клетка, короткие руки, поясничный лордоз[2].
Часто аномалии мочевыделительной системы. В литературе описаны случаи сочетания карциномы печени и данного синдрома. Интеллект обычно нормальный[2].
Диагностика
Основывается на характерной клинической симптоматике[2].
Лечение
Симптоматическое[2].
Примечания
- ↑ Симптомы и синдромы в эндокринологии / Под ред. Ю. И. Караченцева. — 1-е изд. — Х.: ООО «С.А.М.», Харьков, 2006. — С. 137-138, 150. — 227 с. — (Справочное пособие). — 1000 экз. — ISBN 978-966-8591-14-3.
- ↑ 1 2 3 4 5 6 Малая энциклопедия врача-эндокринолога / Под ред. А. С. Ефимова. — 1-е изд. — К.: Медкнига, ДСГ Лтд, Киев, 2007. — С. 335-336. — 360 с. — («Библиотечка практикующего врача»). — 5000 экз. — ISBN 966-7013-23-5.
- ↑ Эндокринология / Под ред. Н. Лавина. — 2-е изд. Пер. с англ. — М.: Практика, 1999. — С. 72, 140. — 1128 с. — 10 000 экз. — ISBN 5-89816-018-3.
Источник