Синдром шильдера аддисона у ребенка

Болезнь Аддисона – заболевание эндокринной системы. Заболевание поражает кору надпочечников, в результате чего развивается хроническая надпочечниковая недостаточность. Случаи заболевания у детей очень редкие, прежде всего в старшей и средней школе. Доказано, что болезнь Аддисона может передаваться наследственным путем.
Кожа и слизистые оболочки усиленно пигментируются, поэтому болезнь в народе называют «бронзовой».
Кожа становится дычато-серого цвета или приобретает цвет загара. Наиболее заметна пигментация в области сосков молочных желез, промежностей, послеоперационных шрамов, подмышек, в полости рта.
Причины
Патогенез болезни Аддисона обусловлен неправильностью регулирования выработки гормонов надпочечниковой корой. Функциональность коры снижается, что побуждает последовательное снижение численности гормонов надпочечников в крови, преимущественно глюкокортикоидов.
[veo class=”veo-yt” string=”DT0fGB9o8z0″]
Определяющие причины развития заболевания:
- На патогенез патологии влияет аутоиммунное поражение надпочечников;
- Возможны случаи туберкулезного поражения надпочечников.
Выделяют и вторичные причины заболевания:
- Хирургическое вмешательство (операция по удалению надпочечников);
- Неверное медикаментозное лечение (при продолжительном курсе терапии лекарствами, блокирующими синтез гормонов коры надпочечников);
- Опухолевые клетки надпочечников или метастазы из близлежащих органов;
- Хронические заболевания воспалительного характера (бластомикоз, амилоидоз).
Симптомы
Надпочечниковая недостаточность сопровождается расстройством всех видов обмена веществ, поэтому симптомы болезни Аддисона очень разнообразные и многочисленные. Хроническая недостаточность развивается медленно, признаки проявляются слабо.
Спустя некоторое время после начального периода развития заболевания проявляются характерные симптомы:
- Анорексия;
- Резкая потеря веса;
- Астения;
- Головокружение;
- Тошнота;
- Рвотные позывы;
- Диарея.
Одновременно у детей наблюдаются вторичные признаки болезни Аддисона:
- Изменение кожных покровов;
- Ребенок может значительно увеличить количество употребляемой соли в пище;
- У детей снижается уровень глюкозы в крови;
- У девочек подросткового возраста возможно нарушение менструального цикла или его приостановление.
Заболеванию свойственны симптомы расстройства сердечно-сосудистой системы:
- Снижается сила и частота сердечных сокращений;
- Нарушается ритм сердечных сокращений;
- Снижается артериальное давление.
У детей наблюдается отставание в росте сердца, что вызывает сердечно-сосудистую недостаточность. Признаки недостаточного кровообращения – отечность нижних конечностей, а также бледность и холодность кожных покровов.
Иногда у больных наблюдается нарушение глотания. Случаются судорожные приступы, как результат нарушения обмена кальция. Ребенок, страдающий болезнью Аддисона, плохо переносит воздействие солнечного излучения.
В большинстве случаев присутствует отставание в развитии ребенка, вызванное слабой выработкой надпочечниками половых гормонов.
О степени тяжести и серьезности поражения надпочечников сигнализируют такие симптомы:
- Мышечная адинамия (уменьшение или абсолютное прекращение двигательных функций из-за мышечной слабости);
- Артериальная гипотензия (снижение уровня артериального давления больше чем на 20% от нормы).
В качестве осложнения у больных иногда развивается аддисонический криз, возникающий на фоне психических или физических травм, операций или инфекционных заболеваний. Клинические симптомы патологии обостряются: прогрессирует снижение давления, усиливается мышечная слабость, ребенок резко теряет массу тела, постоянно присутствует рвота, боль в животе, понос, затруднение мочеиспускания.
При стертых или атипичных формах болезни Аддисона симптомы могут иметь слабо выраженный характер или вовсе отсутствуют. Единственная возможность поставить диагноз в таком случае – лабораторное исследование.
Диагностика
Диагностика заболевания чаще всего проводится на прогрессирующих стадиях развития, поскольку пациенты обращаются к специалистам в том случае, если симптомы становятся более выраженными. На начальных стадиях течения болезни даже лабораторные анализы не дают возможности поставить верный диагноз.
Диагностика при подозрении на болезнь Аддисона включает в себя следующие исследования и анализы:
- Сбор анамнеза. Эндокринолог осматривает ребенка и проводит опрос родителей о возможной генетической наследственности заболевания.
- Стимуляция адренокортикотропного гормона (АКТГ). Тест применяется при диагностике болезни Кушинга и Аддисона. Диагностика позволяет измерить кортизол в плазме до и после инъекции АКТГ. Надпочечниковая недостаточность устанавливается при отсутствии или слабой реакции уровня кортизола на стимуляцию.
- Лабораторные анализы крови и мочи. Исследование применяется с целью выявления в крови недостатка кортикостероидов (основной показатель – гормон кортизол), а также низкого содержания натрия или высокого уровня калия. Недостаточность функционирования почек определяют по уровню азота и креатинина в мочевине. Общий анализ мочи помогает установить диагноз, если у пациента фиксируют сниженную концентрацию обмена продуктов тестостерона.
- Визуальные тесты. При необходимости, врач назначает компьютерную томографию брюшной полости, чтобы оценить размеры надпочечников. Вторичная недостаточность коры надпочечников у детей диагностируется с помощью МРТ гипофиза.
Если существуют сомнения в верности диагноза, используют водную пробу (тест Робинсон-Поуэр Кепплера). Суть исследования – измерение и сравнение нескольких проб мочи в течение пары суток. Образцы мочи хранятся раздельно. Перед проведением теста пациенту назначается безсолевое питание. Тест также анализирует увеличение отделения хлоридов при умеренной задержке мочевины.
В качестве дополнительного теста определяют мочекислый-креатининовый индекс, являющийся отношением количества испускаемой с водой мочевой кислоты к выделяемому креатинину. Индекс, превышающий отметку 0,26, свидетельствует об опухоли коры надпочечников.
Лечение
Лечение заболевания должно быть комплексным, включающим в себя несколько направлений. Разработанные методики терапии позволяют значительно улучшить самочувствие пациента и предупредить возможные последствия. Лечение проводится в амбулаторном режиме под постоянным наблюдением эндокринолога и фтизиатра. Если у ребенка тяжелая форма заболевания, полагается переведение на инвалидность.
Медикаментозное
Базовое лечение направлено на возмещение в организме необходимого уровня кортизола, альдостерона и других гормонов, вырабатываемых надпочечниками. Так называемое заместительное лечение осуществляется с помощью применения глюкокортикоидов или минералокортикоидов синтетической природы.
Гормон кортизол влияет на метаболические процессы, кровяное давление и на функциональность иммунной системы. Кортизол возмещают за счет назначения кортикостероидов (Дексаметазона, Гидрокортизона, Преднизолона).
Доза приема зависит от назначенного препарата (от 1 до 3 раз в сутки). Препарат Кортизол является наиболее популярным в медицинской практике. Необходимость суточной дозы для каждого пациента предусматривается исходя из индивидуальных показателей.
Минералокортикоиды помогают заменить гормон альдостерон. С этой целью применяют Флудрокортизона ацетат. Пациенты с диагнозом «вторичная недостаточность» не нуждаются в замещении альдостерона, поскольку их надпочечники еще способны вырабатывать этот гормон.
При аддисоническом кризе больному необходимо внутривенное введение инъекций стероидов с одновременным уколом раствора глюкозы. При туберкулезном поражении надпочечников назначают противотуберкулезные препараты под строгим присмотром медицинского персонала.
Решение о снижении дозы гормонов или прекращение их применения принимает только врач, поскольку это может спровоцировать нежелательные последствия (например, острую надпочечниковую недостаточность).
Диета
Лечение болезни обязательно предусматривает диету больного. Рацион должен быть насыщен белками, жирами и углеводами. Врачи рекомендуют отдать предпочтение овощам и фруктам, содержащим витамины группы С и В, а именно – увеличить употребление яблок, капусты, лука, яичного белка, фасоли, печени и моркови.
Диета ограничивает количество ионов калия в приеме пище. Поэтому пациентам следует воздержаться от употребления бананов, горохов, картофеля и орехов.
Профилактика
Профилактика не дает возможности избежать заболевания, но способствует улучшению качества жизни ребенка и снижению риска осложнений.
Врачи рекомендуют детям и их родителям придерживаться основных мер профилактики:
- Правильно составленный рацион;
- Активный образ жизни;
- Отсутствие нервных и психических напряжений;
- Детям, которым назначена инвалидность, следует отказаться от тяжелых физических нагрузок;
- Профилактическое медикаментозное лечение;
- Актуальное и адекватное лечение болезней инфекционной природы;
- Крепкий иммунитет;
- Регулярный контроль здоровья у врача.
Оцените статью: 89 Пожалуйста оцените статью
Сейчас на статью оставлено число отзывов: 89 , средняя оценка: 4,08 из 5
Загрузка…
Источник
Лейкодистрофия меланокожная — болезнь Аддисона—Шильдера
Синонимы: Х-сцепленная адренолейкодистрофия, болезнь Аддисона—Шильдера.
Определение. Дегенеративное заболевание белого вещества головного мозга, связанное с недостаточностью надпочечников и с возможным появлением гиперпигментации кожи.
Этиология и патогенез. Тип наследования рецессивный, сцепленный с Х-хромосомой. Это заболевание, обусловленное энзиматическим дефектом обмена жирных кислот. У пациентов постоянно происходит накопление жирных кислот в головном мозге и коре надпочечников.
Отмечаются мутации гена ABCD1. Ген картирован в терминальном сегменте длинного плеча Х-хромосомы, локус Xq28. Ген ABCD1 кодирует трансмембранный белок ALDP, который относится к семейству трансмембранных белков-транспортеров, характеризующихся наличием АТФ-связывающей кассеты.
Мутационное изменение ALDP нарушает транспорт жирных кислот в пероксисомы. Основным пусковым механизмом патогенеза на молекулярном уровне является нарушение деградации насыщенных жирных кислот, поступающих с пищей и синтезируемых в организме de novo в пероксисомах.
Клиника. На основании клинических проявлений, возраста дебюта, скорости прогрессирования неврологических симптомов заболевание подразделяют на 8 фенотипов: детский, юношеский, взрослый, адреномиелоневропатия, изолированная надпочечниковая недостаточность, бессимптомная форма при наличии биохимического дефекта, атипичная форма, симптоматическая Х-адренолейкодистрофия у гетерозигот.
Гиперпигментация кожи чаще всего обнаруживается при детской церебральной форме заболевания и адреномиелоневропатии. Детская церебральная форма приходится на 7,2 +1,7 лет. В 86% случаев неврологические и психические расстройства часто предшествуют клиническим и лабораторным признакам надпочечниковой недостаточности.
Наиболее часто основными симптомами в этом возрасте являются: гиперактивное или, наоборот, аутистическое поведение, эпизоды агрессивности, проблемы обучения, снижение памяти, дефицит внимания, прогрессирующая деменция и нарушение походки. Менее частыми симптомами являются нарушения зрения и слуха, признаки надпочечниковой недостаточности.
По мере прогрессирования заболевания развивается спастический тетрапарез, слепота, глухота, судороги, не отвечающие на антиэпилептическую терапию. Также начальным и облигатным симптомом при этой форме заболевания является гиперпигментация кожи. При осмотре детей с данной болезнью обращают на себя внимание смуглый цвет кожи всего туловища и лица, но особенно гиперпигментация шейных складок, области локтевых и коленных суставов (так называемый симптом «грязных» локтей и коленей), ягодиц, «бронзовые» ладошки.
Адреномиелоневропатия является наиболее частой формой заболевания у взрослых. Возраст начала болезни колеблется в широком диапазоне от 12 до 50 лет. Гиперпигментация кожных покровов иногда манифестирует в раннем детстве, на много лет опережая появление надпочечниковой недостаточности и неврологических расстройств. В 40% случаев надпочечниковая недостаточность предшествует или совпадает с неврологической симптоматикой.
При неврологическом исследовании на начальных этапах выявляют нижний парапарез, снижение вибрационной чувствительности. По мере прогрессирования заболевания развивается спастический тетрапарез, нарушение всех видов чувствительности и функции тазовых органов. У большинства пациентов со временем присоединяются психические нарушения в виде эмоциональных и депрессивных расстройств. Продолжительность жизни очень широко варьирует и иногда вообще не изменена.
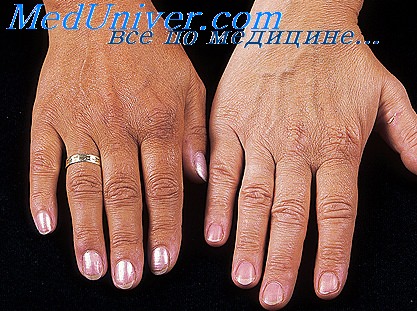
Кожа руки при надпочечниковой недостаточности (болезни Аддисона) слева
— Также рекомендуем «Пигментные пятна (меланозы) при беременности»
Оглавление темы «Гиперпигментации кожи — меланозы»:
- Синдром Вестерхофа — клиника, диагностика
- Синдром Лешке — клиника, диагностика
- Синдром Сильвера-Рассела — клиника, диагностика
- Синдром Февра-Лангепен — клиника, диагностика
- Кофейные пятна при туберозном склерозе
- Изменения кожи при болезни Аддисона
- Изменения кожи при синдроме Нельсона
- Изменения кожи при заболеваниях щитовидной железы (тиреотоксикозе)
- Лейкодистрофия меланокожная — болезнь Аддисона—Шильдера
- Пигментные пятна (меланозы) при беременности
Источник

Адренолейкодистрофия – это наследственная патология из группы пероксисомных болезней, связанная с накоплением в организме жирных кислот с очень длинной углеродной цепью. Заболеванию свойственен клинический полиморфизм – различные формы характеризуются поражением мозговой ткани и надпочечников, проявляясь сочетанием неврологических расстройств (сенсомоторных, эмоционально-когнитивных, поведенческих) и гипокортицизма. Диагностируют патологию по клиническим данным, подтверждая биохимическими, молекулярно-генетическими тестами, МРТ и КТ мозга. Комплексное лечение предполагает диету, фармакотерапию, трансплантацию гемопоэтических клеток.
Общие сведения
Адренолейкодистофия (болезнь Зиммерлинга-Крейтцфельдта, Аддисона-Шильдера) была впервые описана немецкими невропатологами Эрнстом Зиммерлингом и Гансом-Герхардом Крейтцфельдтом в 1923 году. Является не настолько редким заболеванием, как считалось ранее – встречается повсеместно, превосходя по распространенности другие пероксисомные болезни. По данным различных исследований, частота данного варианта лейкодистрофии варьируется от 1:100000 до 1:15000, патология затрагивает представителей всех возрастных групп (детей, подростков, взрослых). Обычно болеют лица мужского пола, женщины являются носителями мутантного гена, но при гетерозиготном генотипе в 20% случаев симптомы обнаруживаются и у них.

Причины адренолейкодистофии
Возникновение патологии связано с мутациями гена ABCD1, занимающего терминальный участок длинного плеча X-хромосомы (локус Xq28). Он кодирует синтез трансмембранного белка-переносчика, называемого адренолейкодистофическим протеином (ALDP). Последний находится на специфических клеточных органеллах, участвующих в реакциях окисления – пероксисомах, отвечая за транспортировку и дальнейшее расщепление очень длинноцепочечных жирных кислот (ОДЦЖК).
Структурный дефект пероксисомного транспортного белка делает его функционально неспособным, что ведет к накоплению в тканях токсических соединений. Уже идентифицировано более 2600 мутаций ABCD1, связанных с заменой нуклеотидов ДНК, потерей локусов, и многие из них вызывают структурные изменения ALDP. Адренолейкодистофии развиваются при наличии в генотипе лишь одного рецессивного гена (у мужчин-гемизигот) или двух его разновидностей (у женщин-гетерозигот).
Патогенез
Из-за структурной аномалии белка-переносчика страдает транспорт ОДЦЖК внутрь пероксисом, где они должны подвергаться β-окислению. В норме насыщенные жирные кислоты с длинной цепью присутствуют в липидах нервной ткани (цереброзидах, сульфатидах), эритроцитах, но при адренолейкодистрофии их содержание может возрастать в тысячу раз. Обычные эфиры холестерина заменяются аномальными с длиной цепи в 24–30 и более атомов углерода. Когда их концентрация в оболочке нервных волокон достигает 10%, миелин дестабилизируется и разрушается.
Накопление ацил-КоА-производных жирных кислот нарушает физико-химические свойства клеточных мембран: повышается проницаемость митохондрий, возрастает концентрация цитозольного кальция. В свою очередь, это приводит к атрофии нейроэндокринной ткани надпочечников. Важным механизмом демиелинизации считают активацию нейроглиальных структур, стимуляцию воспалительных процессов с участием цитокинов (фактора некроза опухолей).
Гистологические изменения при церебральных вариантах адренолейкодистофии характеризуются резким снижением содержания миелина, периваскулярной лимфоцитарно-макрофагальной инфильтрацией. В основном демиелинизирующий процесс начинается с мозолистого тела, постепенно переходя на белое вещество затылочно-теменных областей. Реже наблюдается вовлечение лобных долей, пирамидного тракта.
Классификация
Адренолейкодистофия характеризуется выраженным фенотипическим полиморфизмом, обусловленным различиями пенетрантности и экспрессивности аномального гена. Учитывая время дебюта, основные проявления, скорость нарастания симптоматики, в современной неврологии различают несколько форм заболевания:
- Церебральная. Среди симптомов превалируют неврологические расстройства. Исходя из того, в каком возрасте началась болезнь, выделяют детскую, ювенильную, взрослую формы. Первая встречается чаще остальных (48%).
- Адреномиелонейропатия. Наиболее распространенный вариант для взрослых пациентов (26% случаев). Характеризуется сочетанием кортикоидной недостаточности и спинномозговых нарушений.
- Изолированная надпочечниковая недостаточность. Обычно протекает без поражения нервной системы. В общей структуре адренолейкодистофии изолированному гипокортицизму отводится десятая часть.
- Бессимптомная. Не имея клинической манифестации, проявляется лишь биохимическим дефектом расщепления молекул ОДЦЖК. Обычно встречается среди родственников пациентов, но может рассматриваться как доклиническая стадия.
- Симптоматическая у гетерозиготных носителей. Одновременное присутствие в генотипе доминантной и рецессивной аллели мутантого гена сопровождается болезнью у женщин среднего возраста. На ее долю приходится менее 1%.
- Атипичная. Характеризуется мозжечковыми нарушениями – изолированными или сочетающимися с недостаточностью надпочечников. Встречается крайне редко.
Симптомы адренолейкодистрофии
Клиническая картина патологии очень вариабельна, что определяется конкретным фенотипическим вариантом. Клиницистам чаще всего приходится сталкиваться с признаками церебральной формы, адреномиелонейропатией, изолированной надпочечниковой недостаточностью.
Церебральная форма
Для всех разновидностей церебральной адренолейкодистрофии характерно быстрое прогрессирование. Пик манифестации детской формы приходится на период между 5 и 10 годами жизни. У большинства пациентов нервно-психические расстройства предшествуют признакам надпочечниковой недостаточности. Характерны расстройства поведения, мышления, двигательной сферы. Дети становятся гиперактивными или аутистичными, эпизодически проявляют агрессию. Возникает дефицит внимания, прогрессирующая деменция, нарушается походка.
Реже в симптоматике адренолейкодистрофии присутствуют зрительные (гемианопсия, агнозия, острая потеря зрения), слуховые нарушения, судорожный синдром. Гипокортицизм проявляется кожной пигментацией, мышечной слабостью, периодическими тошнотой и рвотой. Прогрессирование патологического процесса сопровождается спастическим тетрапарезом, слепотой и глухотой, возникают резистентные к медикаментозной терапии судороги. Смертельный исход наступает в сроки от года до 15 лет с момента начала заболевания.
Ювенильная адренолейкодистрофия манифестирует в возрасте 10–21 года. По симптоматике она напоминает детскую форму. У взрослых 30–50 лет патология начинается шизофреноподобным синдромом, нарастающей деменцией. Среди церебральных симптомов присутствуют дисфагия, зрительные нарушения (скотомы). Патология может быстро прогрессировать, но описывают и так называемые хронические варианты, когда процесс на многие годы приостанавливается, а после ремиссии наблюдается внезапное ухудшение с нарастанием неврологического дефицита.
Адреномиелонейропатия
Начинается заболевание в широком возрастном диапазоне – от 12 до 50 лет (чаще между вторым и четвертым десятилетиями жизни). Во многих случаях симптомы хронического гипокортицизма предшествуют или сопутствуют неврологическим расстройствам. Иногда самым первым признаком, возникающим задолго до развертывания всей клинической картины (еще в раннем детстве), становится изолированная гиперпигментация кожи.
Начальные неврологические признаки представлены миелопатией со снижением глубокой чувствительности, нижним парапарезом. Дальнейшее развитие такой адренолейкодистрофии проявляется тетрапарезом с нарушением функции тазовых органов (мочеиспускания, дефекации, эрекции). Со временем присоединяются психические расстройства (депрессия), гипогонадизм, алопеция. Патологическое состояние характеризуется медленным течением, но неуклонно прогрессирует.
Изолированная недостаточность надпочечников
Поражение коры надпочечных желез проявляется сначала глюкокортикоидной, а затем и минералокортикоидной недостаточностью, дебютирует в различные сроки, начиная с двухлетнего возраста. Наиболее частые симптомы представлены потерей аппетита, мышечной слабостью, рвотой. Пациенты теряют в весе, страдают от абдоминальных болей, гипотонии. Кожная гиперпигментация возникает не всегда. Неврологическое исследование указывает на снижение вибрационной чувствительности, гиперрефлексию, интеллектуальные расстройства, возникающие спустя несколько лет.
Симптоматическая адренолейкодистрофия
У некоторых женщин-носителей отмечается возникновение неврологических симптомов без сопутствующих эндокринных сдвигов. Заболевание дебютирует позже, чем у мужчин со спинальным поражением – к 50–60 годам. В тяжелых случаях симптоматика схожа с церебральной формой, умеренные нарушения напоминают адреномиелонейропатию. Наиболее часто выявляют сенсорную атаксию, умеренный спастический парапарез и боли в нижних конечностях, дисфункцию органов малого таза. Недостаточность коры надпочечников возникает редко.
Осложнения
Прогрессирование церебральных форм и адреномиелонейропатии сопровождается выраженным неврологическим дефицитом с инвалидностью. Опасным осложнением гипокортицизма является острая декомпенсация с развитием аддисонического криза, проявляющегося дегидратацией, сердечно-сосудистой, почечной недостаточностью. Быстропрогрессирующие варианты без активной коррекции заканчиваются коматозным состоянием и смертью.
Диагностика
Предположить заболевание удается при внимательной оценке анамнестической информации (наличия семейных случаев, времени манифестации, характера течения), клинических данных. Но этиопатогенетические особенности адренолейкодистрофии устанавливаются при комплексном лабораторно-инструментальном исследовании. Диагностическая программа включает следующие методы:
- Биохимические анализы крови. Обнаружение методом масс-спектрометрии в плазме или клетках (эритроцитах, лейкоцитах, фибробластах кожи) ОДЦЖК – докозановой (C22), тетракозановой (C24), гексакозановой (C26) и их соотношений – рассматривается как способ ранней диагностики (скрининга). Из других биохимических показателей обязательно исследуют уровни кортизола, АКТГ, электролиты.
- Молекулярно-генетические тесты. Для выявления генных мутаций применяют метод прямого секвенирования. Патология характеризуется высокой частотой уникальных мутаций (точечных, сдвига рамки считывания). Корреляцию результатов молекулярно-генетического исследования с тяжестью болезни установить не удается.
- Томография ЦНС. У лиц с церебральными формами МРТ выявляет симметричное снижение интенсивности сигнала в участках мозолистого тела, кортикоспинального тракта с переходом на затылочно-теменную зону. Прогрессирование адренолейкодистрофии сопровождается накоплением контрастного вещества в очагах демиелинизации. На КТ обнаруживают ослабление интенсивности белого мозгового вещества, кальцификаты.
- ЭФИ. Пациентам с адреномиелонейропатией может выполняться электронейромиография, которая выявляет снижение амплитуды моторных и сенсорных ответов. Замедление скорости проведения импульса регистрируют при анализе стволовых вызванных потенциалов (зрительных, слуховых, соматосенсорных).
Дифференцировать заболевание врачу-неврологу приходится со многими состояниями. Церебральные нарушения требуют исключения других лейкодистрофий, рассеянного склероза, подострого склерозирующего энцефалита. Адреномиелонейропатию следует отличать от бокового амиотрофического склероза, фуникулярного миелоза, спинальных опухолей. Дифференцировать изолированный гипокортицизм необходимо с болезнью Аддисона, синдромом Оллгрова.
Лечение адренолейкодистрофии
Консервативная терапия
Выбор обоснованной патогенетической терапии – наиболее острая проблема, стоящая перед клиницистом при лечении конкретного пациента. Поскольку диагностировать заболевание можно еще до развития клинических признаков, особое значение приобретает пресимптоматическая коррекция. При проведении терапии адренолейкодистрофии задействуют консервативные методы:
- Диетотерапию. Для нормализации уровня ОДЦЖК крови рекомендуют придерживаться низкожировой диеты, употреблять масло Лоренцо. Последнее представляет собой смесь триглицеридов олеиновой и эруковой кислот в соотношении 4:1. Диетотерапия показала эффективность лишь на доклинической стадии болезни.
- Фармакотерапию. При явлениях гипокортицизма однозначно показана заместительная терапия глюкококортикоидами и минералокортикоидами, но эти медикаменты никак не влияют на патологический процесс в центральной нервной системе. Симптоматическое лечение миелопатии проводится с помощью нейрометаболических средств, миорелаксантов, витаминов.
- Трансплантацию костного мозга. На ранних стадиях адренолейкодистрофии в детском и подростковом возрасте аллогенная трансплантация гемопоэтических стволовых клеток способна остановить прогрессирование демиелинизации, поэтому признана основным методом лечения. Эффективность процедуры объясняется обновлением нейроглии с расщеплением ОДЦЖК периваскулярными макрофагами.
- Физическую реабилитацию. Методы физического воздействия (иглорефлексотерапия, трансвертебральная микрополяризация) могут быть рекомендованы мужчинам, страдающим адреномиелонейропатией. Они дополняются массажем, лечебной гимнастикой.
Экспериментальное лечение
Несмотря на некоторые успехи, заболевание плохо поддается лечению. Но возрастающая активность исследователей в сфере молекулярной генетики и лучшее понимание патогенеза адренолейкодистрофии поддерживают попытки разработать новые эффективные методы коррекции. Среди них особого внимания заслуживают следующие:
- Трансплантация генно-модифицируемых клеток. Генную терапию считают наиболее перспективным направлением патогенетической коррекции. Она предполагает инъекцию аутологичных стволовых гемопоэтических клеток, в которых с помощью лентивируса проведено восстановление дефектного гена (препарат Lenti-D). Как показывают исследования, это позволяет достичь стабилизация состояния у 88% пациентов.
- Усиление экспрессии генов. Фармакологическая генная коррекция выбрана в качестве альтернативной лечебной стратегии. Путем стимуляции синтеза белка ALDRP (на 66% идентичного мутантному), можно компенсировать дефицит пероксисомного транспортера в популяции фибробластов.
- Иммунотерапия. Выраженность демиелинизации, как предполагается, коррелирует с воспалительной реакцией, опосредованной пока неизвестными иммунными механизмами. Поэтому вариантом лечения рассматривают использование γ-интерферона, иммуноглобулинов, иммуносупрессоров (циклофосфамида, циклоспорина). Но их эффективность пока не подтвердилась.
- Гиполипидемические средства. Препараты, снижающие уровень холестерина, могут нормализовать содержание длинноцепочных молекул в фибробластах. Многообещающим является применение ловастатина, исследования эффективности которого все еще продолжаются. Клинические испытания проходит и препарат собетиром, уже продемонстрировавший способность снижать концентрацию ОДЦЖК в головном мозге, надпочечниках и крови мышей, лишенных гена ABCD1.
Прогноз и профилактика
Долгосрочный прогноз при Х-сцепленной адренолейкодистрофии зависит от конкретного фенотипа. У детей церебральная форма принимает особо тяжелое, быстропрогрессирующее течение с пятилетней выживаемостью на уровне 59%, многие умирают в течение нескольких лет после дебюта болезни. Другие формы могут не влиять на продолжительность жизни, но снижать ее качество за счет потери трудоспособности. Учитывая высокую степень полиморфизма, даже у членов одной семьи прогноз может существенно варьироваться.
Мероприятия первичной профилактики при адренолейкодистрофии предполагают медико-генетическое консультирование вероятных носителей, пренатальную диагностику (биопсию ворсин хориона, анализ амниотической жидкости). Раннее выявление биохимических изменений в крови важно для подтверждения диагноза на доклинических стадиях. В сочетании с активной патогенетической терапией, это поможет избежать прогрессирования, снижая риск дальнейшей инвалидизации.
Источник