Синдром романо уорда у детей

Синдром удлиненного QT (LQT): причины, диагностика, лечениеЭтиология и встречаемость синдрома удлиненного QT. Синдромы удлиненного QT (LQT) — разнородная панэтническая группа нарушений, получивших название каналопатий, поскольку они вызываются дефектами в ионных каналах сердца. Распространеность синдромов удлиненного QT приблизительно 1 на 5000-7000 человек. Большинство случаев удлиненного QT вызвано мутациями в пяти известных генах ионных каналов сердца (KCNQ1, KCNH2, SCN5A, KCNE1.KKCNE2). Генетика, лежащая в основе синдромов удлиненного QT, сложна. Во-первых, существует локусная гетерогенность. Наиболее частый из синдромов удлиненного QT, аутосомно-доминантный синдром Романо-Уорда (MIM №192500), вызван преимущественно мутациями в двух локусах, KCNQ1 и KCNH2, а также содействующим третьим локусом, SCN5A. Во-вторых, разные мутантные аллели в одном и том же локусе могут вызывать два различающихся синдрома удлиненного QT, синдром Романо-Уорда и аутосомно-рецессивный синдром Джервелла-Ланге-Нильсена (MIM №220400). Патогенез синдрома удлиненного QTСиндром удлиненного QT вызывается дефектами реполяризации в клетках сердца. Реполяризация — управляемый процесс, требующий баланса между направленным внутрь клетки потоком натрия и кальция и из клетки — калия. Дисбаланс удлиняет или укорачивает длительность потенциала действия, вызывающего соответственно удлинение или сокращение интервала QT на электрокардиограмме. Большинство случаев синдрома удлиненного QT вызваны мутациями с утратой функции в генах, кодирующих субъединицы или полные белки каналов калия (названия этих генов начинаются с KCN). Эти мутации уменьшают реполяризацию, тем самым продлевая потенциал действия клетки и уменьшая порог для последующей деполяризации. У других пациентов с синдромом удлиненного QT мутации с усилением функции в гене натриевого канала, SCN5A, ведут к повышенному притоку натрия, вызывая аналогичные изменения потенциала действия и эффекты реполяризации.
Фенотип и развитие синдрома удлиненного QTСиндромы удлиненного QT характеризуются удлинением интервала QT и аномалиями зубца Т на электрокардиограмме, включая тахиаритмию и полиморфную желудочковую тахикардию. Желудочковая тахикардия характеризуется изменением амплитуды и скручиванием комплекса QRS. Полиморфная желудочковая тахикардия связана с удлиненным интервалом QT и обычно заканчивается спонтанно, но может упорствовать и прогрессировать в фибрилляцию желудочков. При самом частом варианте синдрома удлиненного QT, Романо-Уорда, обмороки из-за аритмии сердца — наиболее частый признак. Если ребенок остается недиагностированным или не получает лечение, синкопальные состояния повторяются и могут быть летальными в 10-15% случаев. Тем не менее от 30 до 50% индивидуумов с синдромом удлиненного QT никогда не имеют синкопальных симптомов. Сердечные эпизоды чаще всего встречаются в возрасте от 9 до 12 лет, уменьшаясь со временем. Эпизоды могут происходить в любом возрасте, если спровоцированы приемом медикаментов, удлиняющих интервал QT. Нефармакологические триггеры сердечных событий при синдроме Романо-Уорда отличаются в зависимости от ответственного гена. Триггеры LQT1 — обычно адренергические стимулы, включая физическую нагрузку и внезапные эмоции (испуг). Лица с LTQ2 находятся в риске как при нагрузке, так и в покое, а также при слуховых стимулах, например звонок будильника или телефона. Пациенты с LQT3 имеют эпизоды с замедлением сердечных показателей в периоды отдыха и сна. Кроме того, 40% случаев LQT1 проявляют себя до 10-летнего возраста; симптоматика появляется до 10 лет жизни только в 10% случаев LTQ2 и крайне редко при LQT3. Синдром LQT5 редкий, о течении и триггерах известно меньше. Синдром удлиненного QT имеет неполную пенетрантность, как с точки зрения электрокардиографических аномалий, так и синкопальных эпизодов. До 30% больных могут иметь интервалы QT, перекрывающиеся с нормальными колебаниями. Варьирующаяся экспрессия заболевания может происходить как внутри семьи, так и между семьями. Из-за неполной пенетрантности для точного диагноза у членов семьи часто необходима нагрузочная электрокардиография. Синдромы удлиненного QT могут сопровождаться дополнительными данными при медицинском осмотре. Например, синдром Джервелла-Ланге-Нильсена (MIM №220400) характеризуется глубокой врожденной нейросенсорной глухотой в сочетании с синдромом удлиненного QT. Это аутосомно-рецессивное заболевание, вызываемое также определенными мутациями в одном из двух генов (KCNQ1 и KCNE1), участвующих в развитии аутосомно-доминантного синдрома Романо-Уорда. Гетерозиготные родственники пациентов с синдромом Джервелла-Ланге-Нильсена не глухие, но имеют 25% риск развития синдрома удлиненного QT. Особенности фенотипических проявлений синдрома удлиненного QT:
Лечение синдрома удлиненного QTЛечение синдрома удлиненного QT направлено на предотвращение синкопальных эпизодов и остановки сердца. Оптимальное лечение зависит от идентификации ответственного в данном случае гена. Например, терапия b-адреноблокаторами до начала симптомов — наиболее эффективный метод при LQT1 и, отчасти, при LQT2, но его эффективность при LQT3 незначительна. При лечении b-адреноблокаторами необходимо тщательно проверять соответствие возрастным дозам, не прерывать прием лекарственных средств. Для больных с брадикардией могут оказаться необходимыми водители ритма; может потребоваться доступ к внешним дефибрилляторам. Имплантируемые кардиовертеры-дефибрилляторы могут быть необходимыми больным с LQT3 или некоторым лицам с синдромом удлиненного QT, для которых проблематична терапия бета-адреноблокаторами, например больным бронхиальной астмой, депрессией или сахарным диабетом, а также пациентам с остановкой сердца в анамнезе. Некоторые лекарства, например антидепрессивный препарат амитриптилин, фенилэфрин и дифенилгидрамин, или противогрибковые лекарства, включая флуконазол и кетоназол, должны быть исключены из-за их действия, удлиняющего интервал QT или повышения симпатико-тонии. Исключают также виды деятельности и спорта, связанные с интенсивной физической нагрузкой и эмоциональным стрессом.
Риски наследования синдрома удлиненного QTЛица с синдромом Романо-Уорда имеют 50% шанс родить ребенка с унаследованными мутациями в гене. Поскольку частота новых мутаций низкая, большинство больных имеют пораженного родителя (хотя, возможно, бессимптомного). Чрезвычайно важны и могут оказаться жизнесохраняющими подробный семейный анамнез и тщательная кардиологическая оценка членов семьи. Риск повторения для сибсов пациентов с синдромом Джервелла-Ланге-Нильсена — 25%, как и ожидается при аутосомно-рецессивном заболевании. Пенетрантность изолированного синдрома удлиненного QT без глухоты для гетерозиготных носителей синдрома Джервелла-Ланге-Нильсена — 25%. Пример синдрома удлиненного QT. А.Б., 30-летняя женщина с синдромом удлиненного QT (LQT), обратилась в генетическую клинику вместе с мужем, поскольку они планируют беременность. Пара хочет знать риск повторения этого заболевания у детей и подходящие методы генетического тестирования и пренатальной диагностики. Женщина также обеспокоена потенциальным влиянием беременности на ее собственное здоровье. Диагноз синдрома LQT установлен в начале третьего десятилетия жизни, когда она проходила обследование после внезапной смерти ее 15-летнего брата. В целом она — здоровый человек с нормальным слухом, отсутствием дисморфических признаков. У нее никогда не было обморочных состояний. Впоследствии электрокардиографические данные подтвердили диагноз синдрома у нее, ее отца и одной из теток по отцу. Молекулярное тестирование выявило миссенс-мутацию в гене KCNH2, ранее описанную в других семьях с синдромом Романо-Уорда, тип LQT2. Первоначально пациентка получала бета-адреноблокаторы, но ее кардиологи решили, что низкая эффективность b-адреноблокаторов при LQT2 и летальный случай у ее брата оправдывают имплантацию кардиовертера-дефибриллятора как ей, так и ее пораженным родственникам. Пациентка — первый человек в ее семье, проходящий генетическое консультирование по синдрому удлиненнго QT. — Также рекомендуем «Синдром Марфана: причины, диагностика, лечение» Оглавление темы «Наследственные болезни»:
|
Источник
Год утверждения 2016
Профессиональные ассоциации:
- Ассоциация детских кардиологов России
- Союз педиатров России
Оглавление
1. Краткая информация
2. Диагностика
3. Лечение
4. Реабилитация
5. Профилактика
6. Дополнительная информация
1. Краткая информация
1.1 Определение
Синдром удлиненного интервала QT (СУИQT) — наследственное заболевание с высоким риском ВСС, характеризующееся увеличением QT, приступами потери сознания на фоне желудочковых аритмий.
1.2 Этиология и патогенез
Синдром обусловлен мутациями в генах, кодирующих альфа- и бета-субъединицы ионных каналов мембраны кардиомиоцита и белки, участвующие в регуляции ионных токов.
Мутации в генах выявляют в 50-75% случаев.
Нарушение функции ион-специфических каналов приводит к изменению скорости ионных токов и увеличению продолжительности потенциала действия.
Врожденные мутации приводят к полной или частичной потере функции пораженного канала.
Удлинение QT — триггерный фактор для жизнеугрожающей полиморфной желудочковой тахикардии типа «пируэт».
Тахикардия типа «пируэт» — частая причина внезапной смерти при СУИQT.
К удлинению интервала QT также приводит симпатический дисбаланс при левосторонней симпатической иннервации сердца.
1.3 Эпидемиология
Распространенность СУИQT — один случай на 2500-3000 новорожденных.
Среди генотипированных больных «немые» мутации — у 10-36%.
1.4 Кодирование по МКБ-10
I 45.8 – Синдром удлиненного интервала QT
Примеры диагнозов:
- Синдром удлиненного интервала QT, первичный, бессинкопальная форма, I молекулярно-генетический вариант.
- Синдром Джервелла-Ланге-Нильсена (синдром удлиненного интервала QT, первичный, синкопальная форма, врожденная нейросенсорная тугоухость III-IV степени).
- Синдром удлиненного интервала QT, первичный, синкопальная форма, II молекулярно-генетический вариант.
1.5 Классификация
Классификация СУИQT включает 15 молекулярно-генетических вариантов.
Клиническая классификация:
- синкопе в сочетании с удлинением интервала QT;
- удлинение интервала QT в отсутствие синкопе;
- синкопе в отсутствие удлинения интервала QT;
- скрытая форма (“form frust”).
2. Диагностика
2.1 Жалобы и анамнез
Спектр клинических проявлений — от полного отсутствия симптомов до синкопальных состояний и внезапной смерти.
Патогномоничны для СУИQT синкопальные состояния, провоцированные физической и/или эмоциональной нагрузкой, резким звуком, плаванием.
Жалобы на сердцебиение перед потерей сознание редки.
Наличие у родственников 1 и 2 степени приступов потери сознания и/или случаев внезапной смерти до 40 лет; удлинение интервала QT на ЭКГ членов семьи и/или с СУИQT у родственника.
2.2 Физикальное обследование
Фенотипические особенности:
- синдром Романо-Уорда
- синдактилия в 100% синдрома Тимоти
- врожденная нейросенсорная тугоухость при синдроме Джервелла-Ланге-Нильсена
- низкий рост и сколиоз, низко посаженные уши, гипертелоризм, дефекты нёба, микрогнатия, клинодактилия, синдактилия при синдроме Андерсена-Тавила.
Синкопальные состояния манифестируют в любом возрасте, неблагоприятный прогноз – первый синкопе до 6 лет.
У мужчин риск первого синкопе выше в детском возрасте и снижается после подросткового периода.
У женщин наибольшая вероятность первого синкопе — в послеродовом периоде.
Геноспецифические провоцирующие синкопе факторы:
- плавание или ныряния при I молекулярно-генетическом варианте;
- резкий звук при II варианте;
- синкопе во сне при III варианте.
У одного пациента могут быть два и более провоцирующих фактора.
Потеря сознания около 1-2 минут, редко до 20 минут.
Иногда обморок сопровождается судорогами и непроизвольным мочеиспусканием.
Синдром Джервелла-Ланге-Нильсена:
- очень тяжёлая форма СУИQT;
- мутация в генах KCNQ1 и KCNE1;
- аутосомно-рецессивный тип наследования;
- манифестация у 15% до 1 года, у 50% – до 3 лет;
- провоцирующий фактор – нагрузка физическая/эмоциональная;
- бета-блокаторы неэффективны в большинстве случаев;
- показана имплантация кардиовертера-дефибриллятора.
Синдром Андерсена-Тавила:
- периодический калийчувствительный паралич в 100%;
- краниофасциальный и скелетный дисморфизм (сколиоз, микрогнатия и т.д.);
- мутации в гене KCNJ2;
- аутосомно-доминантный тип наследования;
- манифестирует до 10 лет или в подростковом возрасте;
Синдром Тимоти:
- полиорганные поражения;
- синдактилия;
- врожденные пороки сердца;
- иммунодефицитные состояния;
- транзиторная гипогликемия;
- когнитивные нарушения и аутизм;
- мутации в гене CACNA1С;
- наследуется по аутосомно-доминантному типу;
- средняя продолжительность жизни около 2,5 лет.
2.3 Лабораторная диагностика
Биохимический анализ крови:
- электролиты;
- активность кардиоспецифических ферментов;
- маркёры воспаления;
- титр антител к структурам сердца.
Оценка гормонального профиля щитовидной железы.
Молекулярно-генетическая верификация.
2.4 Инструментальная диагностика
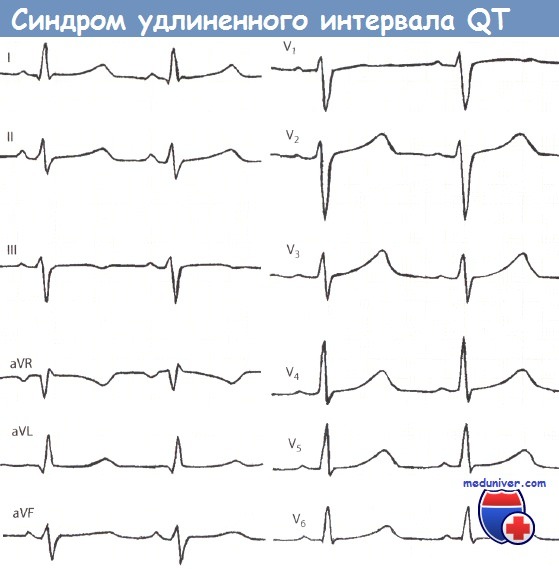
ЭКГ
Критерии P. Schwartz (1993г.), усовершенствованные в 2011 году.
Поверхностная ЭКГ в 12 отведениях в клиноположении, ортоположении и после 10 приседаний со скоростью 50 мм/с.
Коррекция интервал QT по отношению к ЧСС по формуле Базетта.
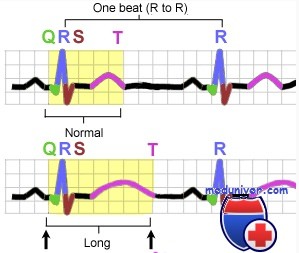
Удлинение интервала QT:
для женщин >460 мс
для мужчин >450 мс.
Дисперсия интервала QT:
у здоровых взрослых от 48±18 до 54±27 мс;
у здоровых детей от 7 до 16 лет 21±11 мс.
Морфология комплекса QRST:
I вариант — зубец Т с широким основанием и косо восходящей элевацией сегмента ST;
II вариант — двугорбый/зазубренный Т в правых грудных отведениях;
III вариант — удлинение QT за счёт сегмента ST, зубец Т обычной морфологии.
Для СУИQT характерна макроальтернация зубца T — изменение полярности и амплитуды в последовательных кардиоциклах.
Альтернация зубца Т ассоциируется с желудочковой тахикардией типа «пируэт» и выраженным более 500 мс удлинением интервала QTс.
Для синдрома Тимоти характерны:
- удлинение QTc до 700 мс;
- развитие функциональной AV-блокады с проведением 2:1;
- макроальтернация зубца Т.
Для синдрома Андерсена-Тавила характерны:
- высокоамплитудные зубцы U;
- Т с покатым, растянутым нисходящим коленом.
Суточное мониторирование ЭКГ (СМЭКГ)
Для выявления маркёров электрической нестабильности миокарда, сопутствующих нарушений ритма и проводимости.
Предпочтительны системы с опцией автоматического анализа интервала QT.
При ХМ-ЭКГ дополнительно оценивают:
- продолжительность интервала QT в автоматическом режиме;
- мануальную оценку интервала QT и QTc на минимальной и максимальной ЧСС;
- особенности морфологии зубца Т;
- наличие зубца U;
- макроальтернации зубца Т.
Эхокардиография (ЭхоКГ)
- Всем больным при первичном обследовании для исключения органической патологии сердца, оценки электромеханического соответствия систолы желудочков на фоне изменения интервала QT.
- Измеряется соотношение времени электрической и механической систолы желудочков.
Тест с физической нагрузкой
- Для дифференциальной диагностики.
- Для определения эффективности антиаритмической терапии.
- Продолжительность интервалов QT и QTc оценивается в исходе, на максимуме нагрузки и на восстановлении.
2.5 Иная диагностика
Дифференциальный диагноз между первичным и вторичным СУИQT, молекулярно-генетическими вариантами синдрома.
3. Лечение
Терапия больных с СУИQT:
- коррекция образа жизни с исключением препаратов, удлиняющих QT;
- медикаментозная и немедикаментозная профилактика ВСС;
- неотложная терапия желудочковой тахикардии типа «пируэт».
Профессиональный спорт противопоказан при синкопальной форме СУИQT и больным из группы высокого риска.
При отсутствии клинических проявлений и генетически подтвержденном СУИQT решение о запрете спорта принимается врачебной комиссией.
3.1 Консервативное лечение
Пожизненная антиаритмическая терапия бета-адреноблокаторами с коррекцией дозы по мере роста пациента.
Бета-адреноблокаторы показаны:
- бессимптомным пациентам с QTc >470 мс;
- пациентам с синкопой;
- при документированной желудочковой тахикардии/фибрилляции желудочков.
Применяются бета-адреноблокаторы:
- неселективный пропранолол0-4.0 мг/кг/сут в 3-4 приёма;
- неселективный нодалол5-1.0 мг/кг/сут в 1-2 приёма;
- селективный атенолол5-2.0 мг/кг/сут в 2 приёма.
Не рекомендуется метопролол, повышающий риск рецидива синкопе.
При III варианте СУИQT и QTc >500 мс с уменьшением QTc более 40 мс после лекарственной пробы вместе с бета-адреноблокатором назначается блокатор натриевых каналов – мексилетин 2.0-5.0 мг/кг на 3 приёма.
3.2 Хирургическое лечение
Имплантация кардиовертера-дефибриллятора для профилактики ВСС показана всем больным:
- перенесшим ВОК;
- при наличии спонтанной устойчивой желудочковой тахикардии с/без синкопе;
- рецидивирующих на фоне антиаритмической терапии синкопе.
Левостороння симпатэктомия рекомендована:
- при сохранении рецидивов желудочковой тахикардии на фоне максимально допустимой дозы бета-блокаторов;
- при противопоказаниях или непереносимости бета-блокаторов;
- при противопоказаниях к имплантации ИКД;
- при отказе от имплантации ИКД.
4. Реабилитация
Медицинской и физической реабилитации больных не требуется.
Детям с частыми срабатываниями ИКД — консультация психолога.
Показано санаторно-курортное лечение в санаториях кардиологического профиля.
5. Профилактика и диспансерное наблюдение
5.1. Профилактика
Для профилактики рецидива желудочковой тахикардии и ВСС необходим динамический контроль факторов риска.
Для своевременной диагностики заболевания обследуют группы риска — членов семьи:
- I и II степени родства больного СУИQT;
- внезапно умерших в молодом возрасте.
5.2. Ведение пациентов
Больные с генетически детерминированными нарушениями ритма наблюдаются в специализированном аритмологическом центре.
Частота посещений зависит от возраста больного и тяжести заболевания.
Контроль эффективности терапии и мониторинг факторов риска:
- при синкопальной форме — 1 раз в 1-6 месяцев;
- без синкопе — не реже 1 раза в год;
- в пубертатном периоде — 1 раз в 6 месяцев.
Первичная госпитализация в специализированное кардиологическое отделение для диагностики и стратификации индивидуального риска ВСС.
Продолжительность госпитализации определяется основным заболеванием.
Контроль системы ИКД у пациентов с имплантированным кардиовертером-дефибриллятором:
- не реже 1 раза в 6 месяцев;
- каждый раз при срабатывании устройства;
- при рецидиве синкопе.
При плановом контроле системы ИКД предварительно выполняют:
- ЭКГ;
- ХМ;
- ЭхоКГ;
- рентгенографию органов грудной клетки в прямой и левой боковой проекциях.
Вакцинация
Решение о вакцинации индивидуальное в зависимости от:
- состояния пациента;
- эффективности медикаментозного контроля аритмии;
- с учётом ранее выявленных провоцирующих факторов.
Детям с синкопальной формой СУИQT вакцинация по индивидуальному графику.
В отсутствие синкопе вакцинация проводится в декретированные сроки.
6. Дополнительная информация, влияющая на течение и исход заболевания
Исходы и прогноз
Прогноз заболевания основывается на риске ВС и зависит от молекулярно-генетического варианта синдрома, возраста манифестации синкопальных состояний, эффективности антиаритмической терапии бета-блокаторами.
Прогноз для жизни благоприятный при регулярном мониторинге факторов риска ВС и своевременной коррекции модифицируемых факторов риска.
Источник