Синдром рихтера при лимфолейкозе лечение

Причины и диагностика синдрома РихтераДолгое время существовали две противоположные концепции на природу синдрома Рихтера. Согласно одной из них, синдром Рихтера представляет собой сочетание двух генетически не связанных друг с другом заболеваний. В клинической практике эта гипотеза находит прямое подтверждение только в тех исключительных случаях, когда течение В-клеточной лимфатической опухоли осложняется присоединением крупноклеточной лимфомы с иным (Т-клеточным) иммунологическим фенотипом. Сложнее трактовка наблюдений, когда обе болезни имеют одинаковую, реже Т-, а чаще В-линейную, принадлежность. В тех случаях, когда на мембране клеток лимфоцитарной и крупноклеточной опухолей обнаруживают иммуноглобулины, идентичные по типу Н- и L-цепей, кажется очевидным, что обе болезни развиваются из одного исходного клона. Иногда экспрессия идентичных по типу L-цепей может сочетаться с разными изотипами (классами) иммуноглобулинов. Такие находки тоже скорее подтверждают, чем опровергают, идею о клональной прогрессии при синдроме Рихтера, поскольку в отличие от лимфоцитарной лимфомы/хронического лимфолейкоза на опухолевых элементах крупноклеточной лимфомы обнаруживались изотипы, свойственные более поздним этапам иммунного ответа —(М + D) => М => G => А (феномен переключения изотипов Н-цепей). Если на мембране лимфоцитов и крупных опухолевых элементов определяются иммуноглобулины одного класса, но с разным типом L-цепей, например Igrvk—IgMA или IgMh—IgMk, предполагают, что заболевания происходят из разных клонов опухолевых клеток. Установлено, что каждая полипептидная цепь иммуноглобулина кодируется несколькими генетическим элементами, которые в зародышевой конфигурации пространственно разобщены. В предшественниках В-лимфоцитов эти элементы в результате рекомбинации ДНК должны расположиться рядом, чтобы образовался единый активный генный комплекс, способный кодировать синтез тяжелых (VHD-JH + СH) или легких (VLJL + CL) полипептидных цепей. Иными словами, в результате соматической перестройки молекулы ДНК происходит объединение рассредоточенных (V1=>n — variable, D1-5 — diversity, J1-4 — joining, Cu,q,y,a,e — constat) генных сегментов, и из множества вариантов производится сборка одного, уникального для данной популяции клеток кло-нального маркера. Этот процесс, обозначаемый как реаранжировка генов иммуноглобулинов, приводит к образованию фрагмента ДНК, отличающегося от зародышевой конфигурации. Образец перестройки (реаранжировки) генов тяжелых и к или X легких цепей является уникальным в каждой конкретной В-клеточной опухоли. В опухоли при этом имеются множественные копии идентичных V(D).1-объединений, отражающих принадлежность клеток к одному клону. При синдроме Рихтера анализ реаранжировки генов иммуноглобулинов методом блоттинга по Саузерну используется как важный дополнительный молекулярно-биологический способ изучения клональной связи между двумя различными по своим морфологическим характеристикам популяциями опухолевых клеток. Метод блот-гибридизации позволяет не только выявить рестрикционные фрагменты ДНК, отражающие генные объединения V(D)J в лимфоцитах крови/костного мозга и клетках экстрамедуллярной опухолевой ткани, но и сравнивать их. Обнаружение одинаковых клональных полос реаранжировки генов Н- и/или L-цепей дает основание полагать, что синдром Рихтера чаще всего представляет собой моноклоновый злокачественный процесс. Обычно в таких случаях обе популяции опухолевых клеток экспрессируют на своей поверхности только один тип L-цепей — либо к, либо X. В тех случаях, когда в лимфоцитах и крупных опухолевых элементах гены иммуноглобулинов перестроены по-разному, утверждается, что заболевания при синдроме Рихтера не имеют клональной связи друг с другом. В этих случаях опухоли, как правило, отличаются по типу синтезируемых L-цепей. Затруднительными для интерпретации представляются ситуации, при которых из двух типов перестройки генов иммуноглобулинов, присутствующих в опухоли, только один совпадает с образцом реаранжировки в В-лимфоцитах крови/костного мозга. Серьезные затруднения в трактовке могут возникать, если результаты генных исследований вступают в противоречие с данными, полученными при иммунологическом фенотипировании опухолевых клеток. Подобная ситуация описана К. Miyamura и соавт.. В публикации речь идет о пациенте 71 года с лимфоцитозом крови/костного мозга, шейной лимфаденопатией, гепато- и спленомегалией и симптомами общей интоксикации. Морфологически в препаратах лимфатического узла обнаружена картина диффузной крупноклеточной лимфомы. В представленном наблюдении клетки лимфоцитарной и крупноклеточной опухолей, с одной стороны, имели идентичный иммунофенотип (CD5+CD19+CD20+HLA-DR+) и одинаковые реаранжировки генов IgH, с другой — они различались по типу экспрессируемых L-цепей — X и к соответственно. Последнее обстоятельство не помешало авторам сделать вывод о том, что обе болезни, вероятнее всего, имели общее клональное происхождение, по крайней мере на начальных этапах малигнизации.
Для теоретического обоснования такого подхода предложены две гипотетические точки зрения. Согласно одной из них, онкогенное событие у данного больного могло произойти очень рано в ряду В-клеточной дифференцировки: после перестройки генов Н-цепей, но до реаранжировки генов L-цепей иммуноглобулинов. Другое объяснение единого происхождения двух заболеваний заключается в том, что исходный (лимфоцитарный) опухолевый клон имел мембранный IgMk. После клональной эволюции в крупноклеточную лимфому делеция к-генов и реаранжировка h-генов произошли только в популяции опухолевых лимфоцитов. Нам представляется вероятным и другое объяснение данного наблюдения: возможно, родоначальные клетки лимфоцитарной опухоли не делетировали к-гены и персистировали на протяжении болезни в «минорных», не улавливаемых методом Саузерна количествах. Длительное время было неясно, насколько лимфоциты лимфоцитарной лимфомы/хронического лимфолейкоза являются функционально активными и способными к дальнейшей дифференцировке. Решению этой проблемы в определенной степени способствовала работа L. F. Bertoli и соавт., в которой, кроме того, продемонстрирован иной подход к изучению клональных взаимоотношений при синдроме Рихтера. Авторы получили моноклональные антитела к идиотипической (антигенной) детерминанте молекул Ig(M+D) Х-типа, экспрессированных на лимфоцитах больной хроническим лимфолейкозом. В двухцветной иммунофлюоресценции В-клетки с мембранным Ig(M+D)A. идентифицировались с помощью антиидиотипических антител как преобладающая в лейкемическом клоне клеточная популяция. Вместе с тем было показано, что в определенной части IgG и IgA В-клеток, а также в большинстве IgG плазмоцитов костного мозга и крови обнаруживалась идиотипическая детерминанта, аналогичная таковой в лейкемических В-лимфоцитах. Следовательно, лейкозные лимфоциты или по крайней мере их некоторая часть способны к восприятию антигенных стимулов, изотипическому переключению и дифференцировке в плазматические клетки. Через 6 лет после установления диагноза хронического лимфолейкоза больная умерла при явлениях дыхательной недостаточности, обусловленной опухолевой инфильтрацией легочной ткани. На вскрытии выявлена диффузная крупноклеточная лимфома, клетки которой экспрессировали на своей поверхности IgMh с таким же идиотипом, как и лимфоциты исходного клона. Кроме того, при блоттинге по Саузерну в лейкемических клетках крови и ткани лимфомы были получены идентичные клональные полосы перестройки генов Н-цепей иммуноглобулинов. Таким образом, в данном наблюдении общее клональное происхождение хронического лимфолейкоза и диффузной крупноклеточной лимфомы было подтверждено не только при изучении иммунофенотипа, генных реаранжировок, но и с помощью антиидиотипических антител к молекулам иммуноглобулинов, экспрессированным на опухолевых клетках. В результате этих исследований одновременно получены косвенные доказательства способности В-лимфоцитов хронического лимфолейкоза к дифференцировке вплоть до плазмоцитов на протяжении самой болезни. При блоке развития лейкемических клеток на более позднем этапе дифференцировки, по-видимому, возможна трансформация в крупноклеточную лимфому. Подобная возможность подтверждается наблюдением Е. Cofrancesco и соавт.. У больной с 6-летним анамнезом хронического лимфолейкоза развилась крупноклеточная лимфома с генерализованным поражением лимфатических узлов, печени, селезенки, кишечника, надпочечников, почек, костей. Обе популяции лимфоидных клеток (мелкие и крупные) имели идентичные реаранжировки генов иммуноглобулинов и сходные иммунофенотипические характеристики (CD5+CD19+CD20+HLA-DR+CD10). Однако крупные клетки лимфомы отличались от лимфоцитов (клеток лимфолейкоза) большей степенью иммунологической дифференцировки в направлении плазмоцитов. Это подтверждалось появлением цитоплазматических IgM, экспрессией CD38, утратой мембранных IgD и снижением розеткоообразования с мышиными эритроцитами. При комбинированных В-клеточных опухолях клональная связь между злокачественными процессами изучали также посредством структурного анализа генов иммуноглобулинов. Работа V. Cherepa-khin и соавт. была первой, в которой по последовательности нуклеотидов ДНК показано, что клетки крупноклеточной лимфомы и хронического лимфолейкоза при синдроме Рихтера могут происходить из одного клона, несмотря на разный иммунофенотип: клетки крупноклеточной опухоли у больного были CD5-негативными. J. Seymour и J. Campbell проанализировали известные исследованные современными методами случаи синдрома Рихтера. Они установили, что примерно в 2/3 случаев хронический лимфолейкоз и развившаяся крупноклеточная лимфома происходят из одного клона, в 1/3 — из разных. Гематологам известно, что далеко не всегда при наличии зрелоклеточной лимфатической опухоли развивается крупноклеточная лимфома. Более того, синдром Рихтера — это редкий клинико-морфологический феномен. По данным различных авторов, он встречается только у 3—10 % больных лимфоцитарной лимфомой/хроническим лимфолейкозом. В такой ситуации понятны попытки выяснить, чем основная масса лимфоцитарных опухолей отличается от той небольшой части, которая осложняется развитием крупноклеточной лимфомы. Выдвигается и обосновывается довольно логичное предположение о возможном существовании подгруппы В-клеточных лимфатических опухолей, более подверженных внешним воздействиям и/или неконтролируемой бласттрансформации. Подтверждением этому может служить обнаружение в лимфоцитах при ХЛЛ, осложненном развитием крупноклеточной лимфомы, замещающей мутации в D- и/или Jн-сегментах генов иммуноглобулинов. Большинство же случаев лимфоцитарной лимфомы/ хронического лимфолейкоза обычно представлено популяцией В-лимфоцитов, не имеющих таких мутаций. Результаты изучения реаранжировки протоонкогенов BCL-1, BCL-2, c-MYC, некоторых супрессорных генов, а также ТР53 иногда представляют большую сложность для интерпретации. Например, при различной реаранжировке генов иммуноглобулинов в опухолевых клетках при лимфолейкозе и крупноклеточной лимфоме могут обнаруживаться идентичные перестройки протоонкогена BCL-2. Пока до конца неясны механизмы развития крупноклеточной опухоли в тех случаях, когда она происходит из того же клеточного клона, что и хронический лимфолейкоз. Изучение влияния ростовых факторов (TGF-p, G-CSF), цитогенетических изменений, мутаций генов ТР53 и р16, чаще наблюдающихся при синдроме Рихтера, чем при ХЛЛ, c-MYC, реаранжировка которого обнаружена в некоторых случаях синдрома Рихтера, не внесло определенности. Много работ посвящено изучению роли вируса Эпштейна — Барр в развитии синдрома Рихтера. Показано, что ДНК вируса обнаружена только при развитии крупноклеточной лимфомы с клетками, напоминающими клетки Рид — Штернберга, и крупноклеточной опухоли с Т-клеточным иммунофенотипом. Во всех этих случаях доказано, что лимфоциты ХЛЛ и клетки лимфомы происходят из разных клонов. Неясной остается роль иммуносупрессии, в частности, вызываемой лечением флударабином. В некоторых сообщениях, основанных на небольшом количестве наблюдений, показано определенное увеличение частоты синдрома Рихтера у больных ХЛЛ, получавших флударабин. В больших сериях, основанных на наблюдениях нескольких сотен длительно прослеженных больных, увеличения синдрома Рихтера у получавших пуриновые аналоги не отмечено. Возможно, указанное некоторыми авторами увеличение частоты крупноклеточной лимфомы при лечении флударабином объясняется более агрессивным течением ХЛЛ у этих больных, что и потребовало применения флударабина. Развитие крупноклеточной лимфомы у больных со зрелоклеточными лимфопролиферативными заболеваниями является плохим в прогностическом отношении признаком и, как правило, сопровождается появлением ряда новых клинических симптомов. Чаще всего наблюдаются следующие: Продолжительность жизни после обнаружения крупноклеточной опухоли обычно колеблется от 6 до 12 мес, несмотря на применение адекватных при лимфомах высокой степени злокачественности методов комбинированной химиотерапии. При синдроме Рихтера иногда наблюдается изолированная экстранодальная локализация очагов крупноклеточной лимфомы. Так, описано поражение кожи, мягких тканей с прорастанием в позвонок и его деструкцией, вещества мозга, яичек, желудка и/или кишечника, бронхиального дерева с эндобронхиальным ростом опухоли. Появление перечисленных признаков у больных с лимфоцитарной опухолью должно служить основанием для проведения диагностической биопсии. Мы наблюдали 13 больных со злокачественными лимфопролиферативными заболеваниями, протекавшими с лимфоцитозом крови и костного мозга, у которых развилась крупноклеточная лимфома. Эти больные составили 3,2 % от общего числа больных с периферическими мелкоклеточными В-лимфомами. Среди больных с синдромом Рихтера было 6 женщин и 7 мужчин в возрасте от 40 до 77 лет. Основным клиническим проявлением злокачественного процесса у 12 больных было увеличение лимфатических узлов разных групп с лимфоцитозом крови и костного мозга. У одного пациента экстрамедуллярный компонент лимфоцитарной опухоли характеризовался изолированным поражением селезенки. В морфологическом субстрате у всех больных преобладали малые лимфоциты. Субстрат лимфоцитарной опухоли характеризовался типичной коэкспрессией зрелыми В-лимфоцитами маркеров CD5 и CD23. Опухолевые элементы крупноклеточной В-лимфомы отличались иммунофенотипически от субстрата лимфоцитарной лимфомы отсутствием экспрессии CD5 во всех, a CD23 — в половине исследованных случаев. Через 8—180 мес (медиана 65 мес) течение зрелоклеточного лимфопролиферативного процесса осложнилось развитием крупноклеточной лимфомы с поражением лимфатических узлов и/или экстранодальной локализацией очагов опухолевого роста. У разных больных наблюдалось поражение различных органов и тканей: кожи, мягких тканей, костей, молочной железы, сальника, плевры с развитием плеврита, носоглотки. Подобная «трансформация» у 8 больных сопровождалась ухудшением состояния, причем у четырех отмечались общие симптомы. У остальных пациентов самочувствие оставалось без изменений. В период развития крупноклеточной (иммунобластной) лимфомы у 5 из 13 больных наблюдалась самопроизвольная регрессия лимфоцито-за крови и костного мозга, т. е. исчезновение основного признака лимфоцитарной опухоли. У 2 больных, напротив, генерализация иммунобластной лим-фомы сопровождалась ростом лимфоцитоза крови и костного мозга до самых высоких значений за весь период наблюдения. Продолжительность жизни после установления диагноза крупноклеточной лимфомы широко варьировала — от 1 до 106 мес (медиана 8 мес). — Также рекомендуем «Лечение синдрома Рихтера — схемы химиотерапии» Оглавление темы «Лейкозы»:
|
Источник

Хронический лимфолейкоз – это онкологическое заболевание, сопровождающееся накоплением атипичных зрелых В-лимфоцитов в периферической крови, печени, селезенке, лимфоузлах и костном мозге. На начальных стадиях проявляется лимфоцитозом и генерализованной лимфоаденопатией. При прогрессировании хронического лимфолейкоза наблюдаются гепатомегалия и спленомегалия, а также анемия и тромбоцитопения, проявляющиеся слабостью, утомляемостью, петехиальными кровоизлияниями и повышенной кровоточивостью. Отмечаются частые инфекции, обусловленные снижением иммунитета. Диагноз устанавливается на основании лабораторных исследований. Лечение – химиотерапия, пересадка костного мозга.
Общие сведения
Хронический лимфолейкоз – заболевание из группы неходжкинских лимфом. Сопровождается увеличением количества морфологически зрелых, но неполноценных В-лимфоцитов. Хронический лимфолейкоз является самой распространенной формой гемобластозов, составляет треть всех лейкозов, диагностируемых в США и странах Европы. Мужчины страдают чаще женщин. Пик заболеваемости приходится на возраст 50-70 лет, в этом периоде выявляется около 70% от общего количества хронических лимфолейкозов.
Пациенты молодого возраста страдают редко, до 40 лет первые симптом болезни возникают всего у 10% больных. В последние годы специалисты отмечают некоторое «омоложение» патологии. Клиническое течение хронического лимфолейкоза очень вариативно, возможно как продолжительное отсутствие прогрессирования, так и крайне агрессивный вариант с летальным исходом в течение 2-3 лет после постановки диагноза. Существует ряд факторов, позволяющих прогнозировать течение заболевания. Лечение осуществляют специалисты в области онкологии и гематологии.
Хронический лимфолейкоз
Причины
Причины возникновения окончательно не выяснены. Хронический лимфолейкоз считается единственным лейкозом с неподтвержденной связью между развитием заболевания и неблагоприятными факторами внешней среды (ионизирующим излучением, контактом с канцерогенными веществами). Специалисты считают, что основным фактором, способствующим развитию хронического лимфолейкоза, является наследственная предрасположенность. Типичные хромосомные мутации, вызывающие повреждения онкогенов на начальной стадии болезни, пока не выявлены, однако исследования подтверждают мутагенную природу заболевания.
Патогенез
Клиническая картина хронического лимфолейкоза обусловлена лимфоцитозом. Причиной лимфоцитоза становится появление большого количества морфологически зрелых, но иммунологически дефектных В-лимфоцитов, неспособных к обеспечению гуморального иммунитета. Ранее считали, что аномальные В-лимфоциты при хроническом лимфолейкозе являются долго живущими клетками и редко подвергаются делению. В последующем эта теория была опровергнута.
Исследования показали, что В-лимфоциты быстро размножаются. Ежедневно в организме больного образуется 0,1-1% от общего количества атипичных клеток. У разных больных поражаются различные клоны клеток, поэтому хронический лимфолейкоз можно рассматривать как группу близкородственных заболеваний с общим этиопатогенезом и сходной клинической симптоматикой.
При изучении клеток выявляется большое разнообразие. В материале могут преобладать широкоплазменные либо узкоплазменные клетки с молодыми либо сморщенными ядрами, почти бесцветной либо ярко окрашенной зернистой цитоплазмой. Пролиферация аномальных клеток происходит в псевдофолликулах – скоплениях лейкозных клеток, располагающихся в лимфоузлах и костном мозге.
Причинами цитопении при хроническом лимфолейкозе являются аутоиммунное разрушение форменных элементов крови и подавление пролиферации стволовых клеток, обусловленное повышением уровня Т-лимфоцитов в селезенке и периферической крови. Кроме того, при наличии киллерных свойств разрушение кровяных клеток могут вызывать атипичные В-лимфоциты.
Классификация
С учетом симптомов, морфологических признаков, скорости прогрессирования и реакции на терапию различают следующие формы болезни:
- Хронический лимфолейкоз с доброкачественным течением. Состояние больного долго остается удовлетворительным. Отмечается медленное увеличение количества лейкоцитов в крови. С момента постановки диагноза до стабильного увеличения лимфоузлов может пройти несколько лет или даже десятилетий. Больные сохраняют трудоспособность и привычный образ жизни.
- Классическая (прогрессирующая) форма хронического лимфолейкоза. Лейкоцитоз нарастает в течение месяцев, а не лет. Отмечается параллельное увеличение лимфоузлов.
- Опухолевая форма хронического лимфолейкоза. Отличительной особенностью этой формы является нерезко выраженный лейкоцитоз при выраженном увеличении лимфоузлов.
- Костномозговая форма хронического лимфолейкоза. Выявляется прогрессирующая цитопения при отсутствии увеличения лимфатических узлов, печени и селезенки.
- Хронический лимфолейкоз с увеличением селезенки.
- Хронический лимфолейкоз с парапротеинемией. Отмечаются симптомы одной из вышеперечисленных форм заболевания в сочетании с моноклональной G- или M-гаммапатией.
- Прелимфоцитарная форма хронического лимфолейкоза. Отличительной особенностью этой формы является наличие лимфоцитов, содержащих нуклеолы, в мазках крови и костного мозга, образцах ткани селезенки и лимфоузлов.
- Волосатоклеточный лейкоз. Выявляются цитопения и спленомегалия при отсутствии увеличения лимфоузлов. При микроскопическом исследовании обнаруживаются лимфоциты с характерным «моложавым» ядром и «неровной» цитоплазмой с обрывами, фестончатыми краями и ростками в виде волосков либо ворсинок.
- Т-клеточная форма хронического лимфолейкоза. Наблюдается в 5% случаев. Сопровождается лейкемической инфильтрацией дермы. Обычно быстро прогрессирует.
Выделяют три стадии клинических стадии хронического лимфолейкоза: начальную, развернутых клинических проявлений и терминальную.
Симптомы хронического лимфолейкоза
На начальной стадии патология протекает бессимптомно и может выявляться только по анализам крови. В течение нескольких месяцев или лет у больного хроническим лимфолейкозом выявляется лимфоцитоз 40-50%. Количество лейкоцитов приближено к верхней границе нормы. В обычном состоянии периферические и висцеральные лимфоузлы не увеличены. В период инфекционных заболеваний лимфатические узлы могут временно увеличиваться, а после выздоровления снова уменьшаться. Первым признаком прогрессирования хронического лимфолейкоза становится стабильное увеличение лимфоузлов, нередко – в сочетании с гепатомегалией и спленомегалией.
Вначале поражаются шейные и подмышечные лимфоузлы, затем – узлы в области средостения и брюшной полости, потом – в паховой области. При пальпации выявляются подвижные безболезненные плотноэластические образования, не спаянные с кожей и близлежащими тканями. Диаметр узлов при хроническом лимфолейкозе может колебаться от 0,5 до 5 и более сантиметров. Крупные периферические лимфоузлы могут выбухать с образованием видимого косметического дефекта. При значительном увеличении печени, селезенки и висцеральных лимфоузлов может наблюдаться сдавление внутренних органов, сопровождающееся различными функциональными нарушениями.
Пациенты с хроническим лимфолейкозом жалуются на слабость, беспричинную утомляемость и снижение трудоспособности. По анализам крови отмечается увеличение лимфоцитоза до 80-90%. Количество эритроцитов и тромбоцитов обычно остается в пределах нормы, у некоторых больных выявляется незначительная тромбоцитопения. На поздних стадиях хронического лимфолейкоза отмечаются снижение веса, ночные поты и повышение температуры до субфебрильных цифр. Характерны расстройства иммунитета. Больные часто страдают простудными заболеваниями, циститом и уретритом. Наблюдается склонность к нагноению ран и частое образование гнойников в подкожной жировой клетчатке.
Причиной летального исхода при хроническом лимфолейкозе часто становятся тяжелые инфекционные заболевания. Возможны воспаления легких, сопровождающиеся спаданием легочной ткани и грубыми нарушениями вентиляции. У некоторых больных развивается экссудативный плеврит, который может осложняться разрывом или сдавлением грудного лимфатического протока. Еще одним частым проявлением развернутого хронического лимфолейкоза является опоясывающий лишай, который в тяжелых случаях становится генерализованным, захватывая всю поверхность кожи, а иногда и слизистые оболочки. Аналогичные поражения могут наблюдаться при герпесе и ветряной оспе.
Осложнения
В числе возможных осложнений хронического лимфолейкоза – инфильтрация преддверно-улиткового нерва, сопровождающаяся расстройствами слуха и шумом в ушах. В терминальной стадии хронического лимфолейкоза может наблюдаться инфильтрация мозговых оболочек, мозгового вещества и нервных корешков. По анализам крови выявляются тромбоцитопения, гемолитическая анемия и гранулоцитопения.
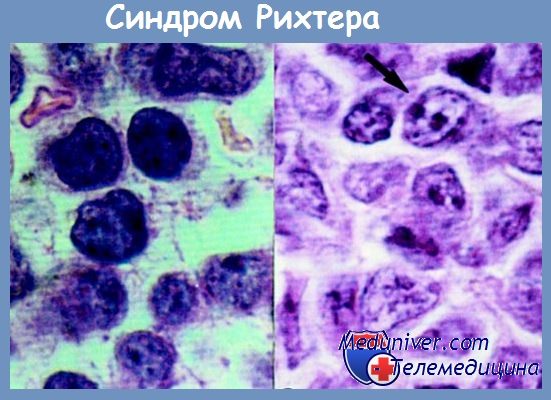
Возможна трансформация хронического лимфолейкоза в синдром Рихтера – диффузную лимфому, проявляющуюся быстрым ростом лимфоузлов и формированием очагов за пределами лимфатической системы. До развития лимфомы доживает около 5% пациентов. В остальных случаях смерть наступает от инфекционных осложнений, кровотечений, анемии и кахексии. У некоторых больных хроническим лимфолейкозом развивается тяжелая почечная недостаточность, обусловленная инфильтрацией почечной паренхимы.
Диагностика
В половине случаев патологию обнаруживают случайно, при обследовании по поводу других заболеваний или при проведении планового осмотра. При постановке диагноза учитывают жалобы, анамнез, данные объективного осмотра, результаты анализов крови и иммунофенотипирования. Диагностическим критерием хронического лимфолейкоза является увеличение количества лейкоцитов в анализе крови до 5×109/л в сочетании с характерными изменениями иммунофенотипа лимфоцитов. При микроскопическом исследовании мазка крови выявляются малые В-лимфоциты и тени Гумпрехта, возможно – в сочетании с атипичными или крупными лимфоцитами. При иммунофенотипировании подтверждается наличие клеток с абберантным иммунофенотипом и клональность.
Определение стадии хронического лимфолейкоза осуществляют на основании клинических проявлений заболевания и результатов объективного осмотра периферических лимфоузлов. Для составления плана лечения и оценки прогноза при хроническом лимфолейкозе проводят цитогенетические исследования. При подозрении на синдром Рихтера назначают биопсию. Для определения причин цитопении выполняют стернальную пункцию костного мозга с последующим микроскопическим исследованием пунктата.
Лечение хронического лимфолейкоза
На начальных стадиях хронического лимфолейкоза применяют выжидательную тактику. Пациентам назначают обследование каждые 3-6 месяцев. При отсутствии признаков прогрессирования ограничиваются наблюдением. Показанием к проведению активного лечения является увеличение количества лейкоцитов вдвое и более в течение полугода. Основным методом лечения хронического лимфолейкоза является химиотерапия. Наиболее эффективной комбинацией лекарственных препаратов обычно становится сочетание ритуксимаба, циклофосфамида и флударабина.
При упорном течении хронического лимфолейкоза назначают большие дозы кортикостероидов, осуществляют пересадку костного мозга. У больных пожилого возраста с тяжелой соматической патологией использование интенсивной химиотерапии и пересадка костного мозга могут быть затруднены. В подобных случаях проводят монохимиотерапию хлорамбуцилом или применяют данный препарат в сочетании с ритуксимабом.
При хроническом лимфолейкозе с аутоиммунной цитопенией назначают преднизолон. Лечение осуществляют до улучшения состояния больного, при этом продолжительность курса терапии составляет не менее 8-12 месяцев. После стабильного улучшения состояния пациента лечение прекращают. Показанием для возобновления терапии является клиническая и лабораторная симптоматика, свидетельствующая о прогрессировании болезни.
Прогноз
Хронический лимфолейкоз рассматривается как практически неизлечимое длительно текущее заболевание с относительно удовлетворительным прогнозом. В 15% случаев наблюдается агрессивное течение с быстрым нарастанием лейкоцитоза и прогрессированием клинической симптоматики. Летальный исход при этой форме хронического лимфолейкоза наступает в течение 2-3 лет. В остальных случаях отмечается медленное прогрессирование, средняя продолжительность жизни с момента постановки диагноза колеблется от 5 до 10 лет. При доброкачественном течении срок жизни может составлять несколько десятилетий. После прохождения курса лечения улучшение наблюдается у 40-70% больных хроническим лимфолейкозом, однако полные ремиссии выявляются редко.
Источник