Синдром прадера вилли и синдром ангельмана

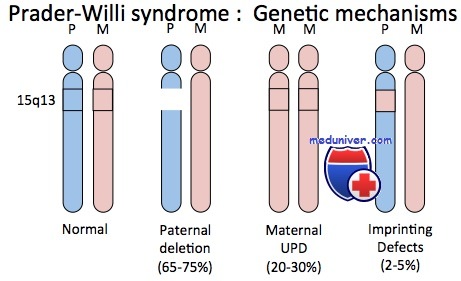
Синдромы Прадера-Вилли и Ангельмана. ХарактеристикаВозможно, наиболее полно изученные примеры роли геномного импринтинга при болезнях человека — синдромы Прадера-Вилли и Ангельмана. Синдром Прадера-Вилли — сравнительно частый дисморфический синдром, характеризующийся ожирением, чрезмерным и беспорядочным аппетитом, небольшими кистями и стопами, низким ростом, гипогонадизмом и умственной отсталостью. Приблизительно в 70% случаев синдрома наблюдают цитогенетическую делецию, затрагивающую проксимальный отдел длинного плеча хромосомы 15 (15q11-q13), причем только в хромосоме, унаследованной от отца больного. Таким образом, геном таких пациентов имеет генетическую информацию в области 15q11-q13, происходящую только от матерей. И наоборот, примерно у 70% пациентов с редким синдромом Ангельмана, характеризующегося необычным лицом, низким ростом, выраженным интеллектуальным отставанием, спастикой и судорогами, отмечают делецию приблизительно той же хромосомной области, но теперь в хромосоме, унаследованной от матери; т.е. пациенты с синдромом Ангельмана имеют генетическую информацию в регионе 15q11-q13, происходящую только от отцов. Эта необычная ситуация удивительным образом доказывает, что родительское происхождение генетического материала в описанных случаях (в хромосоме 15) имеет выраженное влияние на клиническое проявление дефекта.
Приблизительно 30% пациентов с синдромом Прадера-Вилли не имеют цитогенетически обнаруживаемых делеций; но у них выявлены две цитогенетически нормальные хромосомы 15, обе унаследованные от матери. Ситуация иллюстрирует однородительскую дисомию — наличие дисомной линии клеток, содержащих две хромосомы или их части, унаследованные от одного родителя. Если оба экземпляра представлены идентичной хромосомой, состояние называют изодисомией; если присутствуют разные гомологи от одного родителя — гетеродисомией. Приблизительно 3-5% пациентов с синдромом Ангельмана также имеют однородительскую дисомию, только с двумя неповрежденными хромосомами 15 отцовского происхождения. Эти пациенты служат дополнительным подтверждением того, что синдромы Прадера-Вилли и Ангельмана — результат потери соответственно отцовского или материнского вклада генов участка 15q11-q13. Кроме хромосомной делеции и однородительской дисомии, несколько пациентов с синдромами Прадера-Вилли и Ангельмана, вероятно, имеют дефект в самом центре импринтинга. В результате не происходит переключения от женского к мужскому импринтингу в сперматогенезе или от мужского к женскому в овогенезе.
Оплодотворение сперматозоидом, несущим аномально персистирующий женский импринтинг, приведет к рождению ребенка с синдромом Прадера-Вилли; оплодотворение яйцеклетки, имеющей несвойственный ей мужской импринтинг, закончится рождением ребенка с синдромом Ангельмана. Наконец, мутации в материнской копии одного гена — убиквитин-протеин лигазы Е6-АР, как оказалось, вызывают синдром Ангельмана. Ген убиквитин-протеин лигазы Е6-АР расположен в области 15q11-q13 и в норме импринтирован, экспрессируется только материнский аллель в центральной нервной системе (ЦНС). Полагают, что крупные материнские делеции области 15q11-q13 и отцовские однородительские дисомии хромосомы 15, наблюдаемые при синдроме Ангельмана, служат причиной заболевания, так как приводят к утрате материнской копии критически важного импринтированого гена. Мутации для одного импринтированного гена при синдроме Прадера-Вилли пока еще не обнаружены. — Вернуться в содержание раздела «генетика» на нашем сайте Оглавление темы «Аномалии хромосом»:
|

Источник
Синдром Прадера-Вилли (PWS) — это редкое генетическое заболевание, вызванное нарушением хромосомы № 15, которое дети наследуют от своего отца. В дополнение к легкой умственной отсталости и маленькому росту, неудовлетворительный голод, который приводит к хроническому перееданию и ожирению, является типичным симптомом. Поскольку это расстройство невозможно излечить, терапия направлена на облегчение симптомов и на облегчение жизни пациентов.
Что такое синдром Прадера-Вилли?
Синдром Прадера-Вилли, также известный как синдром PWS, является редким генетическим заболеванием. Это заболевание характеризуется недостаточной функцией гипоталамуса (промежуточные участки) и оказывает негативное влияние на правильное развитие нерва. Это затрагивает приблизительно 0,003 — 0,01% населения, и девочки и мальчики затронуты одинаково . По сообщениям, в мире в настоящее время около 400 000 пациентов, которые сталкиваются с этой болезнью
Редкое заболевание было впервые описано в 1956 году швейцарскими врачами Андреа Прадером, Генрихом Вилли и Алексисом Лабхартом , согласно которому синдром также назван сегодня. Двадцать пять лет спустя ген, ответственный за заболевание, был локализован, что значительно упростило диагностику. В настоящее время это заболевание можно обнаружить с помощью генетического тестирования.
Причины PWS
Синдром Прадера-Вилли является первым описанным заболеванием, вызванным генетическим импринтингом. Это связано с потерей экспрессии генов в области 15-й хромосомы (15q11-13), которую плод наследует от своего отца. Это может быть связано с:
- Микроделеция — критическая часть родительской 15-й хромосомы отсутствует (70% случаев)
- Дефект у родителей — обе хромосомы № 15 были унаследованы ребенком (25% случаев)
- Мутации PWS / AS на наследуемой от отца хромосоме (5% случаев)
- Транслокация части 15-й хромосомы (описаны минимальные случаи)
Вероятность того, что генетическое заболевание также возникает у родного брата больного пациента, зависит, прежде всего, от конкретной причины заболевания . Если область 15-й хромосомы полностью отсутствует или монопарентная дисомия, риск относительно невелик. Тем не менее, в случае хромосомной транслокации есть вероятность 25%, что это заболевание затронет другого ребенка, а при мутации PWS / AS — даже 50%.
Каковы симптомы синдрома Прадера-Вилли?
Что касается типичных симптомов, это генетическое расстройство у новорожденных проявляется неразвитым рефлексом всасывания и мышечной слабостью (гипотония). Другие симптомы включают косоглазие, неспособность преуспеть, усталость, апатию или слабый плач. В детстве пациенты маленького роста, и их лица приобретают определенные характеристики, такие как миндалевидные глаза, удлиненные головы или провисающие углы.
Синдром Прадера-Вилли является наиболее распространенной генетической причиной ожирения . Отсутствие аппетита и низкое потребление пищи скоро сменится ненасытным голодом , который никогда не исчезнет полностью. Причиной этой проблемы является высокий уровень грелина, гормона, ответственного за аппетит, который стимулирует чувство голода. Неконтролируемое желание пищи впоследствии приводит к перееданию и ожирению .
ДРУГИЕ ТИПИЧНЫЕ СИМПТОМЫ:
- Задержка двигательного и речевого развития
- Маленькие руки и ноги
- Легкая умственная отсталость
- Нарушения обучения
- Гипогонадизм — снижение производства гонадотропина
- Снижение производства половых гормонов
- Позднее наступление половой зрелости и малых половых органов
- Отсутствие вторичных половых признаков
- Сколиоз
Пациенты с синдромом Прадера-Вилли также характеризуются поведенческими расстройствами . Они могут проявлять упрямство, агрессию или суровое вранье, а также неконтролируемые приступы гнева, которые обычно связаны с лишением пищи, также распространены. ОКР (обсессивно-компульсивное расстройство) также типично . У пациентов возникают проблемы с повторяющимися мыслями или накоплением предметов, и даже незначительное самоповреждение не редкость.
Возможные осложнения
Гипогонадизм и ожирение могут вызвать серьезные осложнения у пациентов с синдромом Прадера-Вилли . Примеры включают в себя:
- Сахарный диабет 2 типа
- Сердечно-сосудистые заболевания
- Инфаркт миокарда
- бесплодие
Диагностика
Врач обычно подозревает, что ребенок страдает от синдрома Прадера-Вилли в соответствии с типичными симптомами, которые сопровождают болезнь. Подобно синдрому Ангелмана (вызванному повреждением 15-й хромосомы от матери), диагноз синдрома Прадера-Вилли можно поставить , проанализировав ДНК крови пациента.
Это генетическое заболевание также можно обнаружить во время беременности, а именно при генетическом исследовании околоплодных вод. Тем не менее, это в первую очередь используется для выявления более частых диагнозов, таких как синдром Дауна или Эдвардса , и, следовательно, PWS обычно более случайный.
Лечение синдрома Прадера-Вилли
Поскольку синдром Прадера-Вилли является результатом генетического расстройства, его невозможно вылечить . Тем не менее, существует ряд способов облегчить типичные симптомы и сделать жизнь пациентов намного проще. Также важным в этом случае является ранняя диагностика, которая поможет родителям хорошо подготовиться к уходу за ребенком с синдромом Прадера-Вилли. Таким образом, они могут заранее удовлетворить его специфические потребности и ознакомиться с важными службами вмешательства.
Сотрудничество нескольких разных специалистов необходимо для успешного управления лечением. Специальный зонд вводится в желудок у новорожденных, у которых есть проблемы с приемом пищи . Однако эта проблема со временем должна исчезнуть, и ребенка можно будет нормально кормить из бутылочки. Если проблемы сохраняются, родители должны проконсультироваться с диетологом. Терапия также включает в себя следующие пункты:
- Лечение ожирения
- Лечение низкого скопления пациентов
- Лечение дефицита половых гормонов
- Профилактика сколиоза
- Овладение мышечной слабостью
- Управление поведенческими расстройствами
ГОРМОНАЛЬНАЯ ТЕРАПИЯ
Частью медикаментозного лечения у пациентов с синдромом Прадера-Вилли является замена гормонов, которые отсутствуют в их организме. Детям дают гормон роста в виде ежедневных подкожных инъекций , которые могут значительно ускорить их рост и оказывают благотворное влияние на неправильный состав тела. Это уменьшает количество подкожного жира, дает ребенку достаточно энергии и даже помогает нормальному развитию лица.
Поскольку у детей с синдромом Прадера-Вилли нарушена гормональная регуляция, также характерна пониженная или полностью отсутствующая активность их половых желез. Терапия, следовательно, состоит из поставки веществ, которые организм не может сделать сам. Использование половых гормонов обычно рекомендуется гинекологом или эндокринологом и должно начинаться в соответствующем возрасте. Девочкам назначают эстроген и прогестерон, а мальчикам — андрогены.
ДИЕТИЧЕСКИЕ РЕКОМЕНДАЦИИ
Мониторинг веса пациентов с синдромом Прадера-Вилли также является одним из наиболее важных поддерживающих методов лечения . Что особенно важно, так это усилия родителей и врачей по предотвращению чрезмерной потери веса , поскольку ребенок не может научиться ограничивать потребление пищи. Люди вокруг него должны быть осведомлены о его диагнозе и уважать его. Другие соответствующие меры включают в себя:
- Акцент на регулярное питание
- Исключение сладких и калорийных продуктов
- Большие порции фруктов и овощей
- Сокращенные порции крахмала
- Храните еду в недоступном для детей месте
- Регулярные упражнения
ХИРУРГИЧЕСКОЕ ЛЕЧЕНИЕ
В некоторых случаях хирургическое лечение также требуется у пациентов с синдромом Прадера-Вилли. Это могут быть:
- Коррекция сколиоза
- Хирургия нисходящих яичек
- Удаление миндалин и миндалин
- Лечение осложнений ожирения
- Лечение абдоминальных событий
ПОДДЕРЖКА ПАЦИЕНТОВ С PWS
Из-за эмоциональных расстройств и снижения интеллекта поддерживающая психологическая терапия всегда должна быть частью лечения синдрома Прадера-Вилли. За больными должен присматривать детский психолог или психиатр, чтобы помочь им справиться с болезнью и ее проявлениями. Сотрудничество с опытными преподавателями также важно.
С помощью родителей, школы, врачей и друзей люди с PWS могут выполнять те же действия, что и их здоровые сверстники . Они могут учиться, заниматься своими хобби, найти работу и стать независимыми. В Чешской Республике родители и пациенты, страдающие синдромом Прадера-Вилли, ассоциируют гражданскую ассоциацию с синдромом Прадера-Вилли.
Источник
Синдром Прадера-Вилли – это редкое генетическое заболевание, характеризующееся грубыми конституциональными нарушениями, когнитивными и психическими расстройствами. Клиническая картина разнообразна, основные симптомы включают ожирение, задержку роста и умственную отсталость. Часто встречается снижение мышечного тонуса, репродуктивная дисфункция. Окончательный диагноз устанавливается на основании молекулярно-генетического исследования. Специфическое лечение не разработано. Осуществляется симптоматическая терапия по основным компонентам синдрома: назначение гипокалорийной диеты и гормональных средств, индивидуальные занятия с дефектологом и т. д.
Общие сведения
Синдром Прадера-Вилли (синдром гипотонии-ожирения) является одной из наиболее выраженных форм генетически обусловленного ожирения. Заболевание впервые было описано в 1956 году швейцарскими педиатрами А. Прадером и Х. Вилли. Несмотря на генетическую природу, болезнь носит спорадический характер. По разным статистическим данным, распространенность синдрома составляет 1:15 000 – 1:25 000 новорожденных. Какие-либо значимые гендерные различия отсутствуют.

Синдром Прадера-Вилли
Причины
Патология развивается в результате мутации 15 хромосомы (сегмента q11.2-q13). Прямой передачи заболевания по наследству не происходит. Хромосомная аномалия возникает в момент оплодотворения яйцеклетки, т. е. обмена родительских генетических материалов. В 65-75% случаев мутация обусловлена дефектом отцовской 15 хромосомы, а в 25-35% – наследованием обеих 15 хромосом от матери. Факторы риска, провоцирующие клинические проявления хромосомной мутации, неизвестны.
Патогенез
Патологические механизмы остаются малоисследованными. Известно, что при этой болезни наблюдается выраженный дисбаланс между процессами липолиза и синтеза жиров в подкожно-жировой клетчатке со сдвигом в сторону последнего. Предполагается, что ведущую роль в ожирении и задержке роста у детей с синдромом Прадера-Вилли играет эндокринная дисрегуляция.
Дисфункция ядер гипоталамуса приводит к снижению выработки многих гормонов, таких как соматотропный гормон, гонадотропины, тиреотропный гормон и пр. Падение концентрации гормона роста и половых гормонов, особенно в детском возрасте, способствует накоплению жировых депо. Характерно повышение уровня пептидного гормона грелина, который является эндогенным стимулятором аппетита.
В генезе нейропсихических расстройств рассматривается роль низкого уровня нейротрофического фактора головного мозга, участвующего в развитии и дифференцировке клеток центральной нервной системы и их функциональной активности. Гипопигментация кожи и волос объясняется подавленной функцией тирозиназы в волосяных фолликулах и меланоцитах.
Симптомы
Клинические проявления начинают манифестировать уже в период внутриутробного развития. Отмечается малая подвижность плода, неправильное предлежание, недоношенность при рождении. Возникает выраженная мышечная гипотония. Значительно ослаблены сосательный и глотательный рефлексы. Это затрудняет кормление ребенка и ведет к недостаточному возрастному набору массы тела. В некоторых случаях необходимо питание через зонд.
Несколько позже присоединяется наиболее характерный симптом – полифагия (патологически повышенный аппетит), вследствие которой ребенок довольно быстро начинает прибавлять в весе, достигая ожирения, вплоть до морбидного. Отложение жира преимущественно происходит в области туловища и проксимальных отделах конечностей.
Выражены нейропсихические нарушения. Речь замедлена, интеллектуальные способности (память, концентрация внимания, последовательная обработка информации) значительно отстают от возрастной нормы. В подростковом периоде нередко наблюдаются обсессивно-компульсивные расстройства, резкие перепады настроения, агрессивное поведение. Из-за недостаточной продукции слюны зубы быстро поражаются кариесом.
Гипогонадизм у мальчиков проявляется гипоплазией мошонки, микропенисом, крипторхизмом, у девочек – недоразвитием половых губ, поздним наступлением менструаций или их полным отсутствием. Возможны нарушения координации, мышечные судороги, косоглазие. Из других конституциональных изменений можно отметить низкий рост, акромикрию (уменьшенный размер кистей и стоп). Типичны гипопигментация кожи, светлые волосы.
Осложнения
Преобладающее число осложнений синдрома Прадера-Вилли связано с морбидным ожирением. Избыток жировой массы способствует раннему развитию инсулинорезистентности, метаболического синдрома и сахарного диабета 2 типа. Нередко встречается неалкогольная жировая болезнь печени (жировой гепатоз). Значительное скопление жира в области шеи обуславливает сужение просвета дыхательных путей.
Вследствие этого более чем у половины пациентов (55-60%) наблюдается синдром обструктивного апноэ сна, который в свою очередь, резко увеличивает риск артериальной гипертензии, инсульта, жизнеугрожающих аритмий. Ожирение также вызывает альвеолярную гиповентиляцию и чрезмерную нагрузку на правые отделы сердца, в результате чего возникает правожелудочковая сердечная недостаточность.
Из-за сниженной минеральной плотности костной ткани любая травма может привести к переломам. Практически все больные страдают первичным бесплодием. Отмечаются частые вирусные инфекции верхних дыхательных путей, бронхиты и пневмонии. Существуют данные о том, что при синдроме ПВ повышается вероятность развития лейкемии и других онкологических заболеваний.
Диагностика
Больных, страдающих синдромом Прадера-Вилли, курируют врачи-педиатры и генетики. При общем осмотре обращают внимание на ослабление мышечного тонуса и сухожильных рефлексов, конституциональные изменения – ожирение, низкий рост. Дополнительное обследование включает следующие исследования:
- Анализы крови. В биохимическом анализе нередко обнаруживается повышение концентрации глюкозы и печеночных трансаминаз (АЛТ, АСТ). Отмечается снижение уровня гонадотропинов (ФСГ, ЛГ), половых гормонов (тестостерона, эстрогенов), соматотропного гормона.
- Денситометрия. При проведении двойной энергетической рентгеновской абсорбциометрии определяются признаки остеопении или остеопороза – показатели плотности костей ниже среднего значения пиковой костной массы более чем на 2,5 SD.
- Определение наличия СОАС. Поскольку обструктивное апноэ представляет угрозу для здоровья и жизни, все пациенты с подозрением на синдром Прадера-Вилли проходят кардиореспираторный мониторинг и полисомнографическое исследование, при которых обнаруживаются высокий индекс дыхательных расстройств и индекс десатурации.
- Генетическое исследование. Выявление микроделеции 15q11-13 с помощью полимеразной цепной реакции, кариотипирования или флуоресцентной гибридизации – основной верифицирующий тест, позволяющий достоверно поставить диагноз.
Дифференциальный диагноз проводится с заболеваниями, которые сопровождаются выраженной мышечной гипотонией и задержкой нейропсихического развития – синдромом Опица-Фриаса, миопатиями, спинальной амиотрофией. Кроме того, синдром ПВ дифференцируется с другими наследственно обусловленными формами ожирения (адипозогенитальная дистрофия, синдром Лоуренса-Муна).
Лечение синдрома Прадера-Вилли
Консервативная терапия
Пациенты подлежат госпитализации в педиатрическое отделение. Эффективные методы этиотропной терапии не разработаны, все лечебные мероприятия носят симптоматический характер. Для борьбы с гипотонией назначаются сеансы массажа и физиотерапевтические методы воздействия. Рекомендуются занятия с логопедом, дефектологом, психотерапевтом. Другие виды лечения синдрома Прадера-Вилли:
- Диета. Основное внимание уделяется изменениям в питании. Необходимо ограничить продукты с высоким содержанием насыщенных жиров и легкоусвояемых углеводов. Общий суточный калораж должен составлять 1000-1200 ккал. Лекарственные препараты, подавляющие аппетит, не используются, так как показали низкую эффективность у больных синдромом ПВ.
- Заместительная гормональная терапия. Рекомендуется подкожное введение рекомбинантного соматотропного гормона даже в раннем детском возрасте еще до наступления ожирения. Для восстановления репродуктивной функции применяются аналоги гонадотропин-рилизинг гормона (гозерелин).
- СИПАП-терапия. Для лечения синдрома обструктивного апноэ наиболее успешным методом является использование специального устройства для автоматической интраназальной вентиляции легких, создающего постоянное положительное давление в верхних дыхательных путях.
- Антиостеопоротическое лечение. При низких показателях плотности костей во избежание патологических переломов назначаются витамин Д (холекальциферол), препараты кальция, бисфосфонаты (золедроновая кислота).
Хирургическое лечение
При наличии определенных показаний (удлиненное мягкое небо, гипертрофия миндалин) для устранения СОАС выполняется хирургическая коррекция – увулопалатофарингопластика, которая заключается в иссечении части мягкого неба, тонзиллэктомии, формировании швов, подтягивающих заднюю стенку глотки. Вероятность рецидива после операции составляет около 50%.
Если не удается добиться снижения массы тела консервативными методами, прибегают к бариатрической хирургии – бандажированию желудка, желудочному шунтированию. Сохранение крипторхизма к концу 1-го года жизни служит показанием к оперативному устранению патологии. Проводится орхипексия – прикрепление яичка к мошонке с помощью швов.
Экспериментальное лечение
Ведутся разработки новых лекарственных средств для терапии синдрома ПВ. Имеются обнадеживающие результаты клинических исследований применения агониста рецепторов окситоцина – карбетоцина. Предлагается воздействовать на кишечную микробиоту больных детей пробиотическими препаратами. В экспериментальных работах на лабораторных животных продемонстрировало лечебный эффект вещество UNC0642, активирующее гены на необходимом участке 15 хромосомы.
Прогноз и профилактика
Продолжительность жизни пациентов, страдающих синдромом ПВ, при своевременной диагностике и адекватном лечении достигает 60-70 лет. В отсутствие превентивных мер смерть может наступить в возрасте 4-5 лет от сердечно-легочной недостаточности. В 50% случаев причиной летального исхода становится обструктивное апноэ сна и вызванные им сердечно-сосудистые катастрофы.
Реже больные погибают от тяжелой респираторной инфекции. Единственным способом предотвращения возникновения заболевания является пренатальная диагностика и прерывание беременности. Основная роль отводится вторичной профилактике – предупреждению осложнений болезни, например вакцинации от гриппа и пневмококковой инфекции.
Источник