Синдром поттера i на узи

Синдромом Поттера называют серьезный дефект развития плода. Характеристику начнем с определения этой аномалии.
Что такое Potter Syndrome?
Синдром Поттера — врожденный дефект развития плода, при котором у него полностью отсутствуют почки, что является следствием уменьшения объема околоплодных жидкостей.
Результатом маловодия является сдавливание плода в утробе матери. У детей также наблюдаются плохо развитые легкие, типичное сморщенное лицо и сдавленный череп, деформированные конечности.
В 1964 году Поттером был описан дефект развития, характеризующийся агенезией (отсутствием развития) обеих почек плода или аплазией (отсутствием какого-либо органа или части тела), сочетающейся с аномалиями лица. Данный синдром упоминается в медицинской литературе так же как синдром Поттера с агенезией 2 почек, почечно-лицевой синдром Гросс, лицевая почечная дисплазия.
Синдром Поттера у плода: причины
Этиология (происхождение) синдрома все еще не изучена до конца. В 50% случаев было выявлено, что первичная аномалия проявляется из-за отсутствия амниотической жидкости (плодных вод) у беременной женщины. Сдавливание плода маткой приводит к развитию аномалий почек, лица (сплющивание и уплощение), сердца, прямой кишки, гениталий, легких (гипоплазия), конечностей (косолапость).

Встречается синдром Поттера у грудничков, детей в возрасте нескольких месяцев, реже — у малышей старше года, причем чаще всего у мальчиков. Частота проявления дефекта: 1 на 50 тыс. родов.
Обследование родителей
Какими симптомами сопровождается синдром Поттера у плода? Большие белые почки — это то, что можно увидеть при ультразвуковом исследовании. Такая агенезия может быть доминантно наследованной (наследственность другого родителя не может ее подавить), т.е. не связанной с количеством околоплодных вод. У одного из родителей может быть недоразвитие или отсутствие одной из почек, что могло быть упущено при медицинских обследованиях ранее.
Агенезия почек наследуется доминантно, а значит, существует 50% вероятность рождения плода, у которого будет наблюдаться синдром Поттера.
Синдром Поттера с 75-процентной вероятностью может проявиться у малыша, если у его родителей обнаружены мутации в гене PKHD1. Они встречаются в среднем у 1 из 50 человек. Подобные мутации не приводят к скорому развитию дефекта у носителя, они могут десятилетиями передаваться из поколения в поколение.
Риск рождения малыша с синдромом Поттера может проявиться при наследовании мутаций от обоих родителей. Если плоду передадутся генетические изменения только отца или матери, то с 50-процентной вероятностью он родится здоровым. 25% малышей вообще не наследуют мутации PKHD1 от обоих родителей, имеющих таковые изменения на генном уровне.
Симптоматика синдрома
Синдром Поттера у плода диагностика выявляет по следующим проявлениям:
- чрезвычайно узкие щели век;
- характерная борозда под линией века;
- недоразвитие нижней челюсти (микрогнация);
- приплюснутый носик;
- аномально большое расстояние между парными органами (в частности глазами) — гипертелоризм;
- выпуклый эпикантус («монгольская складка») — специальная складка у внутреннего уголка глаза, прикрывающая слезный бугорок;
- аномальной формы мягкие большие уши.
Возможность лечения
При рождении ребенка главный симптом — тяжелая дыхательная недостаточность, которая проявляется с первых минут самостоятельной жизни. Проведение искусственной вентиляции легких осложняется пневмотораксом (наличием пузырьков воздуха в плевральной области, которые затрудняют ритмичное движение легких).
К сожалению, подавляющая часть новорожденных, у которых диагностирован синдром Поттера, погибают через несколько часов после родов. При более легких его формах, касающихся дигестивного (пищеварительного) тракта — мочеполовая клоака (соединение анального и мочеполового протока в один), отсутствие перфорации ануса (правильного оформления) — применяется хирургическое вмешательство.
Выживший ребенок с синдромом Поттера
В 2013 году в СМИ появилась информация о новорожденной девочке Эбигейл Бутлер, дочери члена Палаты Представителей парламента Соединенных Штатов, республиканской конгрессвумен Хайме Эррер-Бутлер. Ребенок смог выжить с этим синдромом, препятствующим нормальному функционированию дыхательной системы.
На 5-м месяце беременности женщина узнала, что у девочки не выделяется моча из-за полного отсутствия почек. Причиной был дефицит околоплодных вод у беременной Хайме Эррер-Бутлер. Несмотря на заключение врачей, объявивших этот случай фатальным, женщина вместе с мужем решила сохранить беременность. Чтобы как-то компенсировать малое количество околоплодных жидкостей, в матку Хайме вводили специальный солевой раствор. Джон Хопкинс — доктор, проводивший эту терапию, не мог стопроцентно гарантировать супругам, что лечение приведет к удовлетворительному результату.
Но 15 июля 2013 года маленькая Эбигейл родилась живой. По воспоминаниям матери, девочка не кричала при рождении, но спустя время заплакала — легкие ребенка функционировали. После рождения она сразу же был переведена на диализ — систему, полностью заменяющую почечные функции, выводящую продукты обмена веществ из организма. Через год она заменяется донорскими почки.
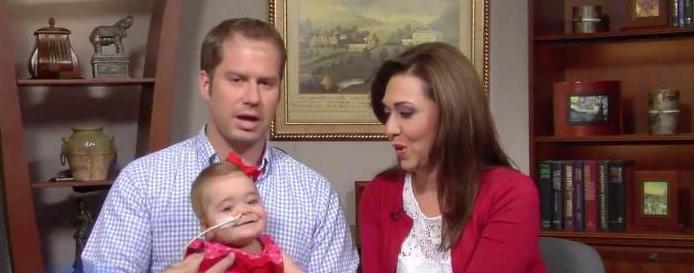
Шанс на рождение живого малыша с синдромом Поттера и его последующей успешной реабилитации в условиях современных реалий пока считается чудом. Поэтому медики настаивают на прерывание беременности при диагностировании у плода такого дефекта. Пример Эбигейл Бутлер не может не обнадеживать, но стоит понимать, что это исключительный случай.
Источник
Поликистозная болезнь почек инфантильного типа (поликистозная болезнь почек I типа синдрома Поттер, аутосомно-рецессивное поликистозное заболевание почек) — двустороннее симметричное увеличение почек вследствие замещения паренхимы вторично расширенными собирательными канальцами без пролиферации соединительной ткани.
Заболевание встречается с частотой 2:110 000 новорожденных и наследуется по аутосомно-рецессивному типу. Основу его составляет первичный дефект собирательных протоков. Почечная лоханка, чашечки и сосочки остаются при этом интактными, так как отсутствует дефект в мочеточниковом зачатке. Сочетанные аномалии встречаются редко.
Диагностика: при ультразвуковом исследовании обнаруживаются двустороннее увеличения почек на фоне маловодия и отсутствие эхотени мочевого пузыря. Типичная гиперэхогенность почек сопровождается повышенной звукопроводимостью вследствие множественных микроскопических кистозных структур в почечной паренхиме.
Прогноз неблагоприятен. Смерть новорожденных наступает от почечной недостаточности.
Акушерская тактика: прерывание беременности в любом сроке.
Поликистозная болезнь почек взрослого типа (аутосомно-доминантная болезнь, гепаторенальная поликистозная болезнь взрослого типа, III тип синдрома Поттер) характеризуется замещением паренхимы почки многочисленными кистами разных размеров, которые образуются вследствие расширения собирательных канальцев и других канальцевых сегментов нефрона. Кисты сосуществуют с интактной почечной тканью. Почки поражены с двух сторон и увеличены, но односторонний процесс может быть первым проявлением заболевания. Вовлечение в процесс печени наблюдается реже и выражается в перипортальном фиброзе, который носит очаговый характер.
Этиология неизвестна. Тип наследования обусловливает 50% риск развития болезни. Ее генетический фокус расположен в 16 паре хромосом.
Частота: один из 1000 человек носит мутантный ген; пенетрация гена происходит в 100% случаев, однако его экспрессивность может варьировать от тяжелых форм, приводящих к смерти в неонатальном периоде, до бессимптомных, обнаруживаемых на аутопсии.
Клинически поликистоз взрослого типа сочетается с кистозными изменениями печени, поджелудочной железы, селезенки, легких, яичников и придатков яичек.
Пренатальная диагностика может быть осуществлена путем биопсии ворсин хориона и исследования биоптата с использованием специального генетического зонда для обнаружения локуса мутантного гена. Описаны единичные наблюдения ультразвуковой диагностики данной патологии.
Прогноз: поликистоз почек относится к хроническим заболеваниям, первые симптомы его могут появиться в любом возрасте. Средний возраст начала клинических проявлений (боли в поясничной области, увеличение почек, почечная недостаточность и уремия) — 35 лет. Гипертензия регистрируется у 50—70% больных.
Акушерская тактика: родители из группы риска должны быть проинформированы о возможности диагностики патологии в I триместре беременности и прерывания беременности в ранние сроки; после достижения плодом жизнеспособности общепринята стандартная акушерская тактика.
Мультикистоз почек (мультикистозная болезнь почек, кистозное заболевание почек II тип синдрома Поттер, диспластическая болезнь почек) — врожденная почечная аномалия, проявляющаяся кистозным перерождением почечной паренхимы вследствие первичного расширения почечных канальцев. Процесс может носить двусторонний, односторонний и сегментарный характер.
Заболевание встречается в основном спорадически и может быть вторичным проявлением в составе других синдромов:
- аутосомно-рецессивных (синдром Меккеля, Денди—Уокера, Робертса, Смита—Лемли—Опица и др.);
- аутосомно-доминантных (синдром Апера);
- хромосомных нарушений.
Двусторонний процесс встречается у одного их 10 000 новорожденных. Односторонний процесс наблюдается в 2 раза чаще у новорожденных мужского пола.
Патогенетические механизмы болезни неизвестны. Предполагают, что эта сложная аномалия может быть результатом двух типов повреждений: недостаточности развития метанефральной бластемы, ответственной за формирование нефронов, и ранней обструктивной уропатии.
Односторонние поражения могут сочетаться с дефектами нервной трубки, диафрагмальной грыжей, расщеплением нёба, аномалиями ЖКТ.
Пренатальная диагностика основана на обнаружении при эхографии кистозно-измененных почек, недостаточной визуализации мочевого пузыря, даже после проведения теста с фуросемидом.
Прогноз: двусторонний процесс, в большинстве наблюдений, приводит к летальному исходу.
Акушерская тактика: при двустороннем процессе, диагностированном в ранние сроки беременности, рекомендуется прерывание беременности; односторонний процесс при нормальном кариотипе и без сочетанных аномалий не влияет на акушерскую тактику. Родоразрешение рекомендуется проводить в перинатальном центре.
Гидронефроз является результатом обструкции мочевыводящих путей в месте соединения почечной лоханки и мочеточника.
Частота его не установлена, так как аномалия представляет спорадический феномен. После рождения в 5 раз чаще диагностируется у лиц мужского пола.
У детей с обструкцией лоханочно-мочеточникового соединения в 27% случаев выявляются сочетанные аномалии мочевыводящих путей, в том числе пузырно-мочеточниковый рефлюкс, двустороннее удвоение мочеточников, двусторонний обструктивный мегауретер, нефункционирующая контралатеральная почка, агенезия контралатеральной почки. В 19% — отмечаются аномалии других органов и систем.
Обструкцию лоханочно-мочеточникового соустья диагностируют с помощью эхографии, основываясь на обнаружении расширения почечной лоханки.
При одностороннем процессе прогноз благоприятный. Акушерская тактика зависит от времени обнаружения патологического процесса и степени нарушение функции почек.
Избранные лекции по акушерству и гинекологии
Под ред. А.Н. Стрижакова, А.И. Давыдова, Л.Д. Белоцерковцевой
Опубликовал Константин Моканов
Источник
синдром Поттера является редким и серьезным заболеванием наследственной аутосомно-рецессивной болезни, которое поражает новорожденных и характеризуется акцентированным олигогидрамниозом (недостатком амниотической жидкости, которая окружает ребенка в эмбриональной стадии), поликистозными почками, почечным агенезом (или отсутствием почка или оба при рождении) и обструктивная уропатия (закупорка мочи).
Эта болезнь была впервые описана патологом Эдит Поттер в 1946 году, который отметил сходные черты лица у серии детей с двусторонним почечным агенезом. Оттуда он постепенно разгадал типичные симптомы болезни.
Это также назвали последовательностью Поттера или последовательностью oligohydramnios. Концепция синдрома Поттера вначале относилась только к случаям, вызванным двусторонним почечным агенезом, хотя в настоящее время многие исследователи используют его для любого случая, который появляется вместе с недостатком амниотической жидкости..
Какова его распространенность?
Синдром Поттера встречается примерно в 1 из каждых 4000 рождений и плодов, однако, есть последние данные, которые оценивают, что частота может быть намного выше.
У мужчин чаще, чем у женщин, развивается этот синдром. Это может быть связано с более высоким показателем у мужчин с брюшной черносливом (или болезнью Игл-Барретта) и обструктивной уропатией (заболевания, связанные с этим синдромом). Хотя подозревалось, что Y-хромосома как-то связана с этим. Во всяком случае, это точно не известно.
Дети, рожденные с этим синдромом, обычно умирают очень рано или рождаются мертвыми. Это обычно происходит из-за легочной недостаточности и двустороннего почечного агенеза.
33% детей умирают в утробе матери, в то время как выживаемость 70% была зарегистрирована у 23 детей с синдромом Поттера и легочной гипоплазией (Klaassen, Neuhaus, Mueller-Wiefel & Kemper, 2007).
У новорожденных с легкой формой синдрома Поттера могут быть осложнения от типичной дыхательной недостаточности, пневмоторакса и острой почечной недостаточности. У тех, кто достигает раннего детства, могут быть хронические заболевания легких и почечная недостаточность.
Как это производится?
Производство мочи у плода является основным механизмом для выработки адекватного объема околоплодных вод, который начинается примерно на четвертом месяце беременности. Плод постоянно глотает амниотическую жидкость, он снова всасывается в кишечнике и затем выводится через почки (через мочу) в амниотическую полость..
При этом заболевании количество околоплодных вод является недостаточным, главным образом потому, что почки ребенка плохо работают. Обычно случается, что в период беременности почки не сформированы должным образом, пропуская одну или обе (почечная агенезия). Хотя также может возникнуть обструкция мочевыводящих путей или, иногда, разрыв мембраны, которая окружает амниотическую жидкость.
Этот недостаток околоплодных вод является основной причиной симптомов синдрома Поттера.
Болезнь Поттера может возникать из-за двух генетических заболеваний, которые являются как аутосомно-доминантным поликистозом почек, так и аутосомно-рецессивным заболеванием. Таким образом, семейная история болезни почек может увеличить риск развития этого синдрома у плода..
Таким образом, в случаях семей с односторонним или двусторонним агенезом почек в анамнезе это может быть аутосомно-доминантным признаком.
Хотя определенные генетические мутации были связаны с состояниями, обычно присутствующими при синдроме Поттера, такими как аутосомно-рецессивный или доминантный поликистоз почек и мультикистозная дисплазия почек; в двустороннем почечном агенезе не найдено ничего определенного.
Таким образом, конкретные генетические признаки в настоящее время точно не известны, и это то, что все еще исследуется.
Что известно, так это то, что, по-видимому, нет прямой связи между злоупотреблением психоактивными веществами или опасными факторами окружающей среды во время беременности при появлении двустороннего почечного агенеза или при синдроме Поттера..
Какие симптомы это имеет??
— Основным дефектом в последовательности Поттера является почечная недостаточность.
— Недостаток амниотической жидкости: это может вызвать много проблем, потому что жидкость помогает смазывать части тела плода, защищает его и способствует развитию легких. Когда эта жидкость отсутствует, амниотическая полость меньше нормальной и в итоге остается мало места для плода, что препятствует нормальному росту.
— Преждевременные роды
— Пороки развития: особенно в нижних конечностях, как в стопах, так и в наклонах ног. Сиреномелия или синдром русалки, который включает в себя слияние ног, также может возникнуть.
— Атипичный внешний вид лица, такой как широкий носовой мост или нос «попугайского клюва», раздельные глаза и уши ниже нормы.
— Избыточная кожа, часто встречающаяся у пораженных кожных складок в области щеки.
— Надпочечники с появлением маленьких овальных дисков, которые давят на заднюю часть живота, связаны с плохой функцией почек.
— Мочевой пузырь меньше нормального и не очень растяжим, хранит очень мало жидкости.
— У мужчин семявыносящий проток и семенные пузырьки могут отсутствовать.
— У женщин матка и верхняя часть влагалища могут не развиваться.
— Анальная атрезия: возникает, когда прямая кишка неправильно соединена с задним проходом. То же самое может произойти в пищеводе, двенадцатиперстной кишке или пупочной артерии.
— Иногда может быть дана врожденная диафрагмальная грыжа, которая препятствует правильному развитию диафрагмы..
— Незрелые легкие или легочная гипоплазия (врожденная аномалия, характеризующаяся прерыванием развития легких согласно Tortajada et al., 2007). Этот механизм не совсем ясен, хотя кажется, что правильное движение амниотической жидкости через легкие во время стадии плода. Очевидно, что если не хватает амниотической жидкости, легкие не будут развиваться должным образом.
— Следовательно, к вышесказанному, существуют серьезные проблемы с дыханием, которые обычно являются причиной ранней смертности среди пострадавших..
Сопутствующие расстройства
В дополнение к уже упомянутым, синдром Поттера был связан с другими проблемами, такими как синдром Дауна, синдром Каллмана и синдром жаберно-ото-почечного (BOR), среди других..
Как это диагностируется?
Во время беременности можно увидеть с помощью ультразвука, если на счету меньше амниотической жидкости, или если у плода есть аномалии в почках или их отсутствие.
Чтобы обнаружить возможные проблемы у новорожденного, может потребоваться сделать рентген легких и брюшной полости.
С другой стороны, вы можете обратиться к генетическому консультанту, который возьмет образец крови у плода для проведения амниоцентеза. Это служит для проверки правильности числа хромосом или изменений в некоторых их частях или транслокациях..
Это может быть полезно для исключения других сопутствующих заболеваний, таких как синдром Дауна. Для выявления возможных наследственных мутаций необходимо изучить геном отца, матери, пострадавшего ребенка и братьев и сестер..
Как вы можете лечить?
Там нет лечения этого заболевания, и его прогноз очень отрицательный, обычно умирает до рождения или вскоре после. Если вы выживаете при рождении, может потребоваться реанимация. Некоторые методы также могут быть использованы для облегчения симптомов и максимально возможного улучшения жизни, таких как трансплантация органов или вмешательство при обструктивной уропатии..
Однако есть случай ребенка с синдромом Поттера, родившегося в июле 2013 года, который был раскрыт Хайме Эррера Бойтлер, которая живет сегодня. Это связано с тем, что за несколько недель до рождения в матку матери вводили физиологический раствор, чтобы помочь легкому развитию плода..
При рождении ребенка было установлено, что вмешательство было успешным и что она могла дышать самостоятельно. Последние наши новости о ней опубликованы 15 апреля 2016 года и выживают после пересадки почки..
ссылки
- От Пьетро М. (19 ноября 2013 г.). Последовательность Олигогидрамниоса (синдром Поттера). Получено от Healthline.
- Гупта С. (21 июня 2015 г.). Синдром Поттера. Получено из Medscape.
- Klaassen I, Neuhaus TJ, Mueller-Wiefel DE, Kemper MJ. Дородовые олигогидрамниозы почечного происхождения: отдаленные результаты. Nephrol Dial трансплантации. 2007 22 февраля (2): 432-9.
- Поттер. (Н.Д.). Получено 24 июня 2016 г. из Википедии.
- Srikanth M. Shastry, S.M., Kolte, S.S. и Санагапати П.Р. (2012). Последовательность Поттера. J Clin Неонатол, 1(3): 157-159.
- Tortajada Girbés, M., Clement Paredes, A., García Muñoz, E., Gracia Antequera, M., Delgado Cordón, F., & Hernández Marco, R. (2007). Детская легочная гипоплазия. Летопись педиатрии, 67: 81-83.
- Weisensee Egan, N. (15 апреля 2016 г.). «Чудо-младенец», принадлежащий конгрессмену, родившийся без почек, наконец-то получает от своего отца: «Мы благословлены».
Источник