Синдром патау по мкб 10
Содержание
- Описание
- Симптомы
- Диагностика
- Лечение
- Прогноз
Названия
Синдром Патау.

Внешний вид ребенка с синдромом Патау (трисомия 13)
Описание
Описан данный синдром в 1960 году, связан с нарушением 13-й хромосомы. Среди новорожденных синдром Патау встречается в соотношении 1:7000. Нет достоверных отличий по частоте встречаемости среди мужского и женского пола. Почти в 50% беременность осложняется многоводием. Масса тела новорожденного значительно ниже нормы при нормальной продолжительности; нередко в родах отмечается асфиксия.
Симптомы
Клиническая картина типична: микроцефальный череп с низким скошенным лбом, наблюдаются вдавленные височные области и дефекты кожи. Обращают на себя узкие глазные щели, расположены горизонтально, характерен глазной гипотелоризм. Отмечается глазная патология: микрофтальмия, колобомы, помутнение роговицы. Нос плоский, широкий, с запавшим переносьем и тупым втянутым кончиком. Низко расположенные ушные раковины сочетаются с маленькими мочками ушей, которые прижаты к голове. Завитки неправильной формы, козелок гипоплазирован. Неправильное развитие кортиева органа нередко обуславливает глухоту.
Одним из частых признаков синдрома является расщелина верхней губы и неба. Со стороны костно-мышечной системы имеют место полидактилия, флексорное положения кистей со своеобразным положением пальцев: 2-4 согнуты, приведены к ладони и перекрыты большим пальцем и мизинцем. Очень часто обнаруживаются пупочные и паховые грыжи, аномалии наружных половых органов: крипторхизм, гипоспадию, гипоплазию мошонки и полового члена у мальчиков, гипертрофию клитора и половых губ у девочек. У детей выявляют глубокую умственную отсталость, у них резко выражено моторное недоразвитие, нередко встречается судорожный синдром.
При патологоанатомическом исследовании обнаруживается множество дефектов развития практически всех систем органов. Масса мозга меньше нормы, недоразвиты или отсутствуют обонятельные луковицы и тракты, мозг часто не разделен на полушария, есть гипоплазия лобных долей и хиазмы зрительного нерва, мозжечка, агенезия мозолистого тела, гидроцефалия. Почти всегда выявляют аномалии сердца и сосудов: нередки дефект межжелудочковой и межпредсердной перегородок, диагностируется незаращение артериального протока, а также патология клапанного аппарата, стеноз легочной артерии. У половины больных с синдромом Патау регистрируют пороки развития почекмочевыводящих путей: кистозная почка, гидронефроз, дисплазия почек, удвоение почек и С такой же частотой встречаются различные дефекты и аномалии расположения органов пищеварения: аномалии поворота кишечника, патология брыжейки, дивертикул Меккеля, изменения поджелудочной железы в виде кистозно-фиброзного процесса. У 50% девочек находят удвоение влагалища и двурогую матку.
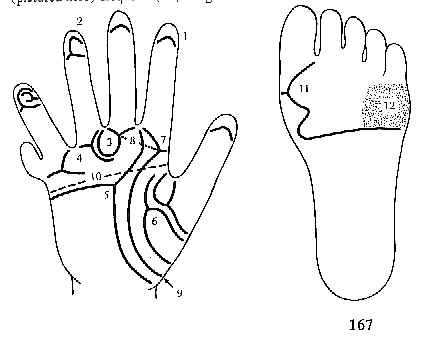
Дерматографические изменения при синдроме Патау (трисомия 13)
Диагностика
Дерматографические изменения заключаются в поперечной ладонной складке, дистальном расположении осевого трирадиуса, повышенной частоте радиальных петель и дуг в основном на большом пальце руки, диагностируется повышение частоты узоров на теноре.
Цитогенетическое исследование выявляет регулярную трисомию хромосомы 13 в 80% случаев. В остальных случаях отмечаются различные формы мозайцизма с участием трисомного клона, транслокации обычного типа t (13qDq). Описанные случаи частичной трисомии 13 чаще являются следствием перицентрической инверсии или сбалансированной транслокации у родителей.
Если при мозайцизме встречаются в основном те же фенотипические признаки, что и при «регулярной» трисомии, то проявления неполной трисомии хромосомы 13 отличаются от типчного синдрома Патау в зависимости от того, какой участок находится в трисомном положении.
Лечение
Лечебная помощь с синдромом Патау неспецифическая: по жизненным показаниям проводят операции по поводу врожденных пороков развития, необходимо общеукрепляющее лечение. Больные дети требуют тщательного ухода, следует проводить профилактику простудных и инфекционных болезней. Дети с синдромом Патау практически всегда имеют глубокую идиотию.
Прогноз
Прогноз для жизни при синдроме Патау неблагоприятный, средняя продолжительность жизни 130 дней: 60% всех больных умирают в первые 3 месяца после рождения, только 10% всех детей живут дольше года.
Источник
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 1 мая 2019;
проверки требуют 7 правок.
Синдром Пата́у (трисомия 13) — генетическое заболевание человека, которое характеризуется возникновением геномной мутации, а именно трисомией по 13-й хромосоме.
История
Трисомия 13 впервые описана Эразмусом Бартолином в 1657 году. Хромосомную природу заболевания выявил доктор Клаус Патау в 1960 году. Заболевание названо в его честь. Синдром Патау также был описан для племён с островов Тихого океана. Считается, что эти случаи были вызваны радиационным заражением, появившимся в результате испытаний ядерного оружия в регионе.
Этиология и эпидемиология
Встречается с частотой 1:7000-1:14000. Имеются два цитогенетических варианта синдрома Патау: простая трисомия и робертсоновская транслокация. Другие цитогенетические варианты (мозаицизм, изохромосома, неробертсоновские транслокации) обнаружены, но они встречаются крайне редко. Клиническая и патологоанатомическая картины простых трисомных форм и транслокационных не различается. 75 % случаев трисомии хромосомы 13 обусловлено появлением дополнительной хромосомы 13. Между частотой возникновения синдрома Патау и возрастом матери прослеживается зависимость, хотя и менее строгая, чем в случае синдрома Дауна. 25 % случаев СП — следствие транслокации с вовлечением хромосом 13-й пары, в том числе в трёх из четырёх таких случаев мутация de novo. В четверти случаев транслокация с вовлечением хромосом 13-й пары имеет наследственный характер с возвратным риском 14 %.
Соотношение полов при синдроме Патау близко к 1:1. Дети с синдромом Патау рождаются с истинной пренатальной гипоплазией (на 25 — 30 % ниже средних величин), которую нельзя объяснить небольшой недоношенностью (средний срок беременности 38,5 недель). Риск возникновения этого синдрома у потомства увеличивается с возрастом матери, достигая пика в среднем к 31 году[2].
Проявления заболевания
Характерным осложнением беременности при вынашивании плода с синдромом Патау является многоводие: оно встречается почти в 50 % случаев Синдрома Патау.
При синдроме Патау наблюдаются тяжёлые врождённые пороки. Дети с синдромом Патау рождаются с массой тела ниже нормы (2500 г). У них выявляются умеренная микроцефалия, нарушение развития различных отделов ЦНС, низкий скошенный лоб, суженные глазные щели, расстояние между которыми уменьшено, микрофтальмия и колобома, помутнение роговицы, запавшая переносица, широкое основание носа, деформированные ушные раковины, расщелина верхней губы и нёба, полидактилия, флексорное положение кистей, короткая шея. У 80 % новорождённых встречаются пороки развития сердца: дефекты межжелудочковой и межпредсердной перегородок, транспозиции сосудов и др. Наблюдаются фиброкистозные изменения поджелудочной железы, добавочные селезёнки, эмбриональная пупочная грыжа. Почки увеличены, имеют повышенную дольчатость и кисты в корковом слое, выявляются пороки развития половых органов. Для СП характерна задержка умственного развития.
В связи с тяжёлыми врождёнными пороками развития большинство детей с синдромом Патау умирают в первые недели или месяцы (95 % — до 1 года).
Однако некоторые больные живут в течение нескольких лет. Более того, в развитых странах отмечается тенденция увеличения продолжительности жизни больных синдромом Патау до 5 лет (около 15 % детей) и даже до 10 лет (2 — 3 % детей).
Оставшиеся в живых страдают глубокой идиотией.
Другие синдромы врождённых пороков развития (синдромы Меккеля и Мора, тригоноцефалия Опитца) по отдельным признакам совпадают с синдромом Патау. Решающим фактором в диагностике является исследование хромосом. Цитогенетическое исследование показано во всех случаях, в том числе у умерших детей. Точный цитогенетический диагноз необходим для прогноза здоровья будущих детей.
Лечение
Исправить хромосомные нарушения невозможно. Комплексная работа группы различных специалистов заключается в постоянном контроле за состоянием здоровья больного и поддержке семьи.
См. также
- Хромосомные заболевания
- Синдром Дауна
- Синдром Эдвардса
Примечания
Ссылки
- https://web.archive.org/web/20070929094344/https://schools.keldysh.ru/school1413/pro_2005/z/hrbol.htm
- https://www.medkurs.ru/lecture2k/genetics/gl19/4288.html
- https://rh-conflict.narod.ru/student/lectures/hrombol.htm
Источник
Рубрика МКБ-10: Q91.7
МКБ-10 / Q00-Q99 КЛАСС XVII Врожденные аномалии пороки развития, деформации и хромосомные нарушения / Q90-Q99 Хромосомные аномалии, не классифицированные в других рубриках / Q91 Синдром Эдвардса и синдром Патау
Определение и общие сведения[править]
Трисомия по 13-й хромосоме
Синонимы: синдром Патау
Трисомия 13 представляет собой хромосомную аномалию, вызванную наличием дополнительной хромосомы 13 и характеризуется пороками развития головного мозга (голопроэнцефалия), лицевым дисморфизмом, глазными аномалиями, постсаксиальными полидактилиями, висцеральными мальформациями (кардиопатия) и тяжелой задержкой психомоторного развития.
Заболеваемость синдромом Патау оценивается в пределах от 1/8000 до 1/15 000 родов.
Этиология и патогенез[править]
Свободная трисомия 13 встречается примерно в 75% случаев. В 20% случаев трисомия 13 связана с транслокацией Робертсона, в которой сверхновая хромосома 13 присоединяется к другой акроцентрической хромосоме (хромосоме 13, 14, 15, 21 или 22). В редких случаях синдромом Патау вызван взаимной транслокацией между хромосомой 13 и неакроцентрической хромосомой. Мозаичная трисомия 13, при которой имеются как трисомные, так и нормальные клетки, сообщалась у нескольких пациентов с клинической картиной, которая варьировалась между нормальным фенотипом и классической трисомией 13 в зависимости от количества трисоматических клеток, присутствующих в тканях.
Клинические проявления[править]
Внутриутробная смертность при синдроме Патау составляет более 95%. Неврологические проявления тяжелые — с гипотонией и гипореактивностью, с очевидным отсутствием осознания окружающих. Голопрозэнцефалия (вызванная дефектом в разделении мозга на два полушария) присутствует в 70% случаев и может проявляться на МРТ в виде слияния полушарий различной степени. Аномалии лицевого черепа могут варьироваться от гипертелоризма и верхнечелюстного агенеза (80% случаев) до цебоцефалии или циклопии с отсутствием скелета носа. Также могут присутствовать расщепление губы/неба, микро- или анофтальмия, колобома (даже при отсутствии серьезных уродств головного мозга), участки затылочной кожной аплазии, постсаксиальная полидактилия, пороки развития сердца (80% случаев) и мочеполовой системы.
Синдром Патау неуточненный: Диагностика[править]
Трисомия 13 может быть заподозрена во время беременности на основнии УЗИ (голопрозэнцефалия, полидактилия) и может быть подтверждена кариотипическим анализом плода.
Дифференциальный диагноз[править]
Синдром Патау неуточненный: Лечение[править]
Лечение только поддерживающее.
Хирургическое лечение пороков развития мало способствует улучшению неблагоприятного прогноза для синдрома Патау: половина младенцев умирает в течение первого месяца жизни, а 90% умирают в возрасте до 1 года от сердечных, почечных или неврологических осложнений. Сообщалось о случаях хорошей выживаемости (в некоторых случаях во взрослую жизнь) в случаях мозаики или частичной трисомии, а также при отсутствии серьезных пороков развития головного мозга. В целом у немозаичных пациентов развивается только ограниченная автономия (отсутствие речи и способности к передвижению).
Профилактика[править]
Риск повторения трисомии (21, 13 или 18) в семьях со случаями синдрома Патау составляет около 1%. Однако в семьях, в которых трисомия 13 связана с транслокацией (робертсоновской или сбалансированной), риск повторения выше, если один из родителей является носителем сбалансированной транслокации.
Прочее[править]
Источники (ссылки)[править]
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник
Утратил силу — Архив
![]()
РЦРЗ (Республиканский центр развития здравоохранения МЗ РК)
Версия: Архив — Клинические протоколы МЗ РК — 2010 (Приказ №239)
Категории МКБ:
Синдром Дауна неуточненный (Q90.9)
Общая информация
Краткое описание
Хромосомные заболевания классифицируются по типам аберраций и вовлеченным хромосомам. Изменение модального числа хромосом проявляется отсутствием какой-либо хромосомы (моносомия) или появлением добавочной хромосомы (трисомия, тетрасомия). Примером таких аномалий являются трисомия 21 (болезнь Дауна), трисомия 18 (синдром Эдварса), трисомия 13 (синдром Патау) и т. д.
Термин «синдром» используется для обозначения устойчивых сочетаний патологических симптомов как уточненной этиологии, так и этиологически неясных. В клинической генетике и тератологии термин «синдром» имеет несколько значений. Он часто применяется как синоним «болезни», когда речь идет о единой нозологической форме с определенной этиологией. При этом синдром чаще называют — врожденные нарушения.
Синдром Дауна — трисомия 21 является самой частой хромосомной патологией у человека. Частота болезни Дауна среди новорожденных составляет 1: 650, среди больных умственной отсталостью болезнь Дауна — самая частая нозологическая форма, она составляет около 10%.
Синдром Эдварса — частота среди новорожденных в среднем составляет 1: 7000; девочки поражаются в 3 с лишним раза чаще, чем мальчики.
Синдром Патау — частота синдрома среди новорожденных в среднем составляет 1:7000. Оба пола поражаются с равной частотой. Масса тела при рождении значительно ниже нормы. Клиническая картина весьма типична.
Протокол «Хромосомные аномалии»
Код по МКБ-10: Q 90; Q 91
Q 90 Синдром Дауна
Q 90.0 Трисомия 21, мейтоническое нерасхождение
Q 90.1 Трисомия 21, мозаицизм (митотическое нерасхождение)
Q 90.9 Синдром Дауна неуточненный
Q 91.0 Трисомия 18,мейтоническое
Q 91.1 Трисомия 18, мозаицизм (митотическое нерасхождение)
Q 91.2 Трисомия 18, транслокация
Q 91.3 Синдром Эдварса неуточненный
Q 91.4 Трисомия 13, мейтоническое нерасхождение
Q 91.5 Трисомия 13, мозаицизм (митотическое нерасхождение)
Q 91.6 Трисомия 13, транслокация
Q 91.7 Синдром Патау
Мобильное приложение «MedElement»
— Профессиональные медицинские справочники. Стандарты лечения
— Коммуникация с пациентами: вопросы, отзывы, запись на прием
Скачать приложение для ANDROID / для iOS
Мобильное приложение «MedElement»
— Профессиональные медицинские справочники
— Коммуникация с пациентами: вопросы, отзывы, запись на прием
Скачать приложение для ANDROID / для iOS
Классификация
1. Мейтоническое нерасхождение.
2. Мозаицизм.
3. Транслокация.
4. Неуточненная форма.
Диагностика
Диагностические критерии
Жалобы и анамнез: задержка в психоречевом развитии, снижение мышления, памяти, внимания, расторможенность, врожденные пороки развития со стороны органов зрения, опорно-двигательного аппарата; отягощенная наследственность, перинатальная патология.
Физикальное обследование
Синдром Дауна (фенотипические проявления) — брахицефальный череп со скошенным затылком и уплощенным лицом, косой разрез глаз (наружный угол выше внутреннего), эпикант, гипертелоризм, расширенное и уплощенное переносье. Маленькие недоразвитые ушные раковины расположены низко, верхняя челюсть недоразвита. Увеличенный «складчатый язык», высокое небо, неправильный рост зубов, короткая шея, широкие кисти с короткими пальцами. В психоневрологическом статусе — мышечная гипотония, недостаточная моторика, интеллектуальная недостаточность.
Синдром Эдварса (фенотипические проявления) — череп долихоцефальный, сдавленный с боков, с низким лбом и широко выступающим затылком; иногда встречается микроцефалия и гидроцефалия. Надглазничные валики сглажены, глазные щели узкие, наблюдается эпикант, гипертелоризм, птоз, часто встречается глазная патология: микрофтальмия, колобома, катаракта. Переносье вдавлено, но спинка носа тонкая, выступающая. Ушные раковины расположены очень низко, почти на уровне углов рта, часто отсутствуют мочка и козелок. Нижняя челюсть маленькая, треугольной формы с короткой верхней губой, нёбо высокое, иногда с расщелиной, шея короткая. Почти всегда можно обнаружить различные аномалии опорно-двигательного аппарата: грудная клетка расширена, грудина укорочена, таз узкий, конечности деформированы, ограничена подвижность в тазобедренных суставах, косолапость, стопы в виде «качалок. Во всех случаях интеллект снижен.
Синдром Патау (фенотипические проявления) — микроцефальный череп с низким скошенным лбом, вдавленными височными областями. Глазные щели узкие, расположены горизонтально, гипотелоризм, почти всегда встречается глазная патология: чаще микрофтальмия или анофтальмия, реже колобомы, помутнение роговицы, циклопия. Нос плоский, широкий, с запавшим переносьем и тупым втянутым кончиком. Ушные раковины расположены очень низко, иногда на уровне рта, маленькие мочки прижаты к голове, завитки неправильной формы, козелок гипоплазирован. Неправильное развитие кортиева органа нередко обусловливает глухоту. Одним из самых частых и демонстративных признаков синдрома являются расщелина мягкого и твердого неба, как двухсторонняя, так и односторонняя, почти всегда расщепление верхней губы сопровождается расщелиной неба. Почти у всех детей имеются аномалии костно-мышечной системы, чаще полидактилия и своеобразное положение кистей. У всех детей отмечается глубокая умственная отсталость.
Лабораторные исследования: кариотип, общий анализ крови и мочи.
Инструментальные исследования:
1. Электроэнцефалография (ЭЭГ): на ЭЭГ задержка формирования возрастной корковой ритмики, диффузные изменения электрогенеза.
2. Электромиография (ЭМГ).
3. Компьютерная томография головного мозга (КТ).
Показания для консультации специалистов:
— логопед;
— психолог;
— ЛОР-сурдолог;
— окулист;
— кардиолог;
— генетик;
— педиатр;
— ортопед.
Минимум обследования при направлении в стационар:
1. Общий анализ крови и мочи.
2. АЛТ.
3. АСТ.
4. Кал на яйца глист.
Основные диагностические мероприятия:
1. Общий анализ крови.
2. Общий анализ мочи.
3. ЭЭГ.
4. Исследование слуха.
5. ЛОР-сурдолог.
6. КТ головного мозга.
7. Логопед.
8. Психолог.
9. Окулист.
10. Кариотип.
11. Генетик.
12. ЭКГ.
13. Кардиолог.
14. ЭхоКГ.
15. УЗИ органов брюшной полости.
Дополнительные диагностические мероприятия:
1. ИФА на токсоплазмоз.
2. ИФА на цитомегаловирус.
3. Анализ мочи на обменные нарушения.
4. Консультация педиатра.
Дифференциальный диагноз
Признак | Синдром Дауна | Синдром Эвдарса | Синдром Патау |
Кариотип | Трисомия хромосомы 21 | Трисомия хромосомы 18 | Трисомия хромосомы 13 |
Форма черепа | Брахицефальный | Долихоцефальный, сдавленный с боков, с низким лбом и широким выступающим затылком, иногда микроцефалия или гидроцефалия | Микроцефальный череп с низким скошенным лбом, вдавленными височными областями |
Глазная патология | Косой разрез глаз, эпикант, пятна Брушфилда (светлые пятна на радужке), гипертелоризм, расширенное и уплощенное переносье | Глазные щели узкие, эпикакант, гипертелоризм, птоз, микрофтальмия, колобома, катаракта | Глазные щели узкие, расположены горизонтально, гипотелоризм, микрофтальмия или анофтальмия, реже колобомы, помутнение роговицы, циклопия |
Опорно-двигательный аппарат | Короткая шея, широкие кисти с короткими пальцами и укороченными искривленными V пальцами (клинодактилия), расширенные промежутки между I и II пальцами стоп. Встречаются и другие скелетные аномалии: синдактилия, деформация грудины — воронкообразная или килевидная грудь, укорочение трубчатых костей | Грудная клетка расширена, грудина укорочена, таз узкий, конечности деформированы, ограничена подвижность в тазобедренных суставах. Кисти и пальцы короткие, V пальцы искривлены, гипоплазированный I палец кисти расположен дистально. Пальцы сжаты в кулак по типу «флексорной аномалии», синдактилия II и III пальцев. Типична для трисомии 18 формы, стопы в виде «качалки», встречается косолапость | Чаще полидактилия и своеобразное положение костей. Флексорное положение кистей сочетается со своеобразным расположением пальцев: II и IV пальцами, стопы часто деформированы |
Онлайн-консультация врача
Посоветоваться с опытным специалистом, не выходя из дома!
- Консультация по вопросам здоровья
- Интерпретация результатов анализов, исследований
- Второе мнение относительно диагноза, лечения
Выбрать врача
Лечение
Тактика лечения
Цели лечения:
— активизация психического развития;
— пополнение пассивного и активного словарного запаса;
— коррекция поведения;
— повышение эмоционального тонуса, настроения ребенка;
— обучение навыкам самообслуживания;
— социальная адаптация.
Немедикаментозное лечение:
— индивидуальные занятия с логопедом;
— занятия с психологом;
— кондуктивная педагогика;
— ЛФК;
— массаж;
— физиолечение.
Медикаментозное лечение
Широко используют в последнее время препараты ноотропного ряда — нейропротекторы, с целью улучшения обменных процессов в головном мозгу. Большинство ноотропных препаратов в связи с их психостимулирующим действием, назначают в первую половину дня. Продолжительность курсов лечения ноотропами составляет от одного до двух-трех месяцев: церебролизин, ампулы 1 мл в/м; пирацетам, ампулы 5 мл 20%; гинкго-билоба (танакан), таблетки 40 мг; пиритинол гидрохлорид (энцефабол), драже 100 мг, суспензия — 5 мл содержит 80,5 мг пиритинола (соотв.100 мг пиритинола гидрохлорида).
Энцефабол — минимум противопоказаний, разрешен к применению с первого года жизни. Дозирование суспензии (с содержанием в 1 мл 20 мг энцефабола) детям 3-5 лет — суточная доза 200-300 мг (12-15 мг массы тела) назначают в 2 приема — утром (после завтрака) и днем (после дневного сна и полдника).
Продолжительность курса 6-12 недель, целесообразен длительный прием, при котором повышается работоспособность и способность к обучению, улучшаются высшие психические функции.
Актовегин, ампулы 2 мл 80 мг, драже-форте 200 мг активного вещества. Нейрометаболический препарат, содержащий исключительно физиологические компоненты. Детям назначается в драже-форте, прием до еды по ½ -1 драже 2-3 раза в день (в зависимости от возраста и выраженности симптомов заболевания) до 17 часов. Продолжительность терапии 1-2 месяца.
Ангиопротекторы с целью улучшения мозгового кровообращения: винпоцетин, циннаризин.
Витамины группы В: В1, В6, В12, нейромультивит — специальный комплекс витаминов группы В с направленным нейротропным действием, неуробекс, фолиевая кислота, аевит.
Седативная терапия по показаниям: ноофен, ново-пассит.
Профилактические мероприятия:
1. Профилактика травматизма.
2. Профилактика вирусных и бактериальных инфекций.
Дальнейшее ведение: регулярные занятия с логопедом, дефектологом, психологом, социальная адаптация ребенка, оформление в специализированный детский сад, детям школьного возраста прохождение медико-педагогической комиссии для решения вопроса об обучении ребенка.
Основные медикаменты:
1. Актовегин, ампулы 2 мл, 80 мг, драже 200 мг
2. Винпоцетин, таблетки 5 мг
3. Пирацетам, ампулы 5 мл 20%
4. Пирацетам, таблетки 0,2 и 0,4
5. Пиридоксин гидрохлорид, ампулы, 1 мл 5%
6. Танакан, таблетки 40 мг
7. Тиамин хлорид, ампулы 5% 1 мл
8. Фолиевая кислота, таблетки 0,001
9. Церебролизин, ампулы 1 мл
10. Цианокобаламин, ампулы 1 мл 200 мкг и 500 мкг
Дополнительные медикаменты:
1. Аевит, капсулы
2. Глицин, таблетки 0,1
3. Гопантеновая кислота (пантокальцин), таблетки 0,25
4. Магния лактат+пиридоксин гидрохлорид, таблетки магне В6
5. Нейромультивит, таблетки
6. Неуробекс, таблетки
7. Ново-пассит, таблетки, раствор
8. Ноофен, таблетки 025
9. Пиритинол, драже 0,1, суспензия
10. Циннаризин, таблетки 25 мг
Индикаторы эффективности лечения:
1. Улучшение внимания, памяти, работоспособности.
2. Пополнение пассивного и активного запаса слов.
3. Повышение эмоционального и психического тонуса.
Госпитализация
Показания к госпитализации (плановая): задержка психоречевого и моторного развития, различные врожденные пороки развития.
Информация
Источники и литература
- Протоколы диагностики и лечения заболеваний МЗ РК (Приказ №239 от 07.04.2010)
- Ю.А. Якунин. Болезни нервной системы у новорожденных и детей раннего возраста. Москва 1979
Справочник детского психиатра и невропатолога. Под редакцией Л.А.Булаховой. Киев 1997
Л.О. Бадалян. Детская неврология. Москва 1998
Г.С.Маринчева, В.И.Гаврилов. Умственная отсталость при наследственных болезнях. Москва 1988
С.К. Козлова, Е. Семанова. Наследственные синдромы и медико-генетическое консультирование. Ленинград 1987
- Ю.А. Якунин. Болезни нервной системы у новорожденных и детей раннего возраста. Москва 1979
Информация
Список разработчиков:
№ | Разработчик | Место работы | Должность |
1. | Кадыржанова Галия Баекеновна | РДКБ «Аксай», психоневрологическое отделение №3 | Заведующая отделением |
2. | Серова Татьяна Константиновна | РДКБ «Аксай», психоневрологическое отделение №1 | Заведующая отделением |
3. | Мухамбетова Гульнара Амерзаевна | Кафедра нервных болезней, КазНМУ | Ассистент, кандидат медицинских наук |
4. | Балбаева Айым Сергазиевна | РДКБ «Аксай», психоневрологическое отделение №3 | Врач-невропатолог |
Прикреплённые файлы
Внимание!
Если вы не являетесь медицинским специалистом:
- Занимаясь самолечением, вы можете нанести непоправимый вред своему здоровью.
- Информация, размещенная на сайте MedElement и в мобильных приложениях «MedElement (МедЭлемент)», «Lekar Pro»,
«Dariger Pro», «Заболевания: справочник терапевта», не может и не должна заменять очную консультацию врача.
Обязательно
обращайтесь в медицинские учреждения при наличии каких-либо заболеваний или беспокоящих вас симптомов.
- Выбор лекарственных средств и их дозировки, должен быть оговорен со специалистом. Только врач может
назначить
нужное лекарство и его дозировку с учетом заболевания и состояния организма больного.
- Сайт MedElement и мобильные приложения «MedElement (МедЭлемент)», «Lekar Pro»,
«Dariger Pro», «Заболевания: справочник терапевта» являются исключительно информационно-справочными ресурсами.
Информация, размещенная на данном
сайте, не должна использоваться для самовольного изменения предписаний врача.
- Редакция MedElement не несет ответственности за какой-либо ущерб здоровью или материальный ущерб, возникший
в
результате использования данного сайта.
Источник