Синдром орбели у ребенка фото

Дети с синдромом Орбели имеют интеллектуальную инвалидность и врожденные пороки развития. Их тяжесть и доступные методы лечения, как диагностируется заболевание при беременности и после рождения, прогноз качества и продолжительности жизни — информация для родителей.
В этой статье:
- Что такое синдром Орбели?
- Фото больных
- Симптомы
- Клинический случай
Что такое синдром Орбели?
Синдром Орбели — это врожденное генетическое заболевание, возникающее при отсутствии (удалении) генетического материала на длинном плече 13 хромосомы. Тяжесть состояния и клиническая картина зависят от количества удаленных генов и их расположения.
Спектр нарушений, характерных для детей с удалением сегмента 13 хромосомы, включает задержку развития, умственную отсталость, поведенческие проблемы и особенности внешности. Болезнь Орбели также называют делецией или моносомией длинного плеча 13 хромосомы.
Тест кариотипа (анализ крови) обоих родителей покажет, была ли делеция унаследована. У большинства родителей детей с синдромом Орбели хромосомная аномалия отсутствует, но у одного из тестируемых может обнаружиться сбалансированная транслокация (разрыв участка хромосомы или присоединение к другой хромосоме с потерей генетического материала или без такого). Сбалансированная транслокация не вызывает симптомов, но увеличивает риск рождения младенца с генетическими аномалиями.
Лечение зависит от признаков синдрома у ребенка и подбирается индивидуально.
Фото больных
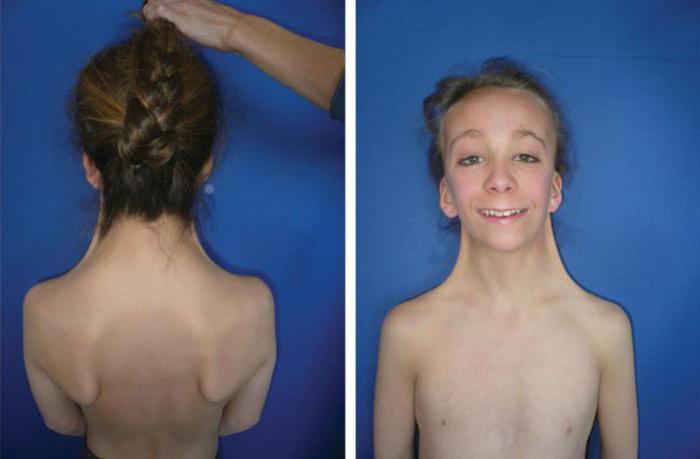
При синдроме Орбели дети имеют специфическую внешность и сочетанные дефекты развития. Фото дают представление о заболевании.
Симптомы
Признаки очевидны при рождении, клиническая картина дополняется по мере взросления. Из первых и самых распространенных симптомов выделятся низкий вес новорожденного, пороки развития черепно-лицевого скелета, глаз, конечностей, половых органов и другие физические аномалии.
У всех детей с диагнозом синдром Орбели наблюдается задержка психомоторного развития: с трудом дается приобретение навыков, требующих умственной и физической активности. Обязательно наличие умственной неполноценности различной степени выраженности.
Задержка роста от умеренной до тяжелой может наблюдаться в любом возрасте или сопровождать весь процесс взросления, что приведет к низкорослости.
Для синдрома Орбели характерны многие аномалии строения лица и черепа (необязательно сопровождают заболевание и сами по себе не являются его признаком):
- микроцефалия (размер мозга ниже нормы, маленькая голова);
- тригоноцефалия (ранее закрытие метопического шва, соединяющего две половины лобной кости, треугольная голова);
- широкий и плоский носовой мост;
- большие, искривленные или низко расположенные уши;
- микрогнатия (недоразвитая, маленькая нижняя челюсть) с аномально выраженной верхней челюстью;
- дистальный прикус (выпирают передние зубы);
- волчья пасть (расщелина неба);
- заячья губа (расщелина верхней губы);
- крыловидная шея (кожные складки с обоих сторон шеи протяженностью до предплечья).
Возможные нарушения строения головного мозга:
- эпилепсия;
- гидроцефалия (скопление спинномозговой жидкости в ликворной системе);
- голопрозэнцефалия (мозг не разделен на полушария);
- менингоцеле (мозговая грыжа);
- отсутствие частей мозга, например, агенезия мозолистого тела или обонятельного отдела;
- гипоплазия (недоразвитие) или агенезия (отсутствие) мозжечка;
- синдром Денди Уокера.
Дефекты развития нервной трубки у плода (при синдроме Орбели встречаются крайне редко, обычно в сочетании с другими генетическими заболеваниями):
- анэнцефалия;
- энцефалоцеле;
- расщепление позвоночника.
Аномалии развития глаза (возможные варианты):
- микрофтальмия (маленький размер и деформация одного или обоих глазных яблок);
- широко посаженные глаза;
- опущение верхнего века;
- эпикантальные (монгольские) складки, закрывающие уголки глаз.
- катаракта;
- помутнение роговицы;
- отсутствие радужной оболочки.
Нарушения строения глаза при синдроме Орбели влияют на зрение, редко вызывают слепоту.
Пороки развития конечностей:
- гипоплазия (недоразвитие) или отсутствие больших пальцев;
- мизинец скошен внутрь;
- слияние пальцев;
- короткие пальцы;
- короткие ноги;
- скошенные стопы (косолапость).
Пороки развития скелета:
- аномалии развития ребер и/или позвоночных костей;
- сколиоз (боковое искривление позвоночника).
Генитальные аномалии часто встречаются у мальчиков и характеризуют синдром Орбели в медицинской литературе:
- недоразвитие пениса;
- смещение отверстия мочеиспускательного канала вниз;
- неопущение яичка в мошонку;
- маленькая мошонка;
- двойная мошонка;
- микропенис.
У девочек возможны неправильное развитие, гипоплазия, отсутствие женских половых органов.
У обоих полов:
- промежностный свищ;
- стеноз или отсутствие анального отверстия.
Аномалии внутренних органов у детей с синдромом Орбели:
- врожденные предсердные и желудочковые дефекты сердца;
- открытое овальное окно в сердце (часто не проявляется, но может спровоцировать худобу, мягкую задержку роста, пониженную сопротивляемость к респираторным инфекциям, одышку, легкую утомляемость при физических нагрузках, аритмию);
- дефект межжелудочковой перегородки (малый может закрыться самостоятельно, умеренный спровоцирует застойную сердечную недостаточность);
- гипоплазия, эктопия, поликистоз или отсутствие почки;
- дефекты легких.
Тяжелые дефекты строения внутренних органов могут привести к смерти новорожденного.
Основные симптомы всех видов частичной делеции длинного плеча 13 хромосомы и их частотность в процентах.
| Признаки | Количество затронутых детей (%) |
|---|---|
| Низкий вес новорожденного (меньше 2,5 кг) | 45 |
| Задержка роста | 30 |
| Психомоторная отсталость | 100 |
| Микроцефалия | 30 |
| Брахицефалия | 75 |
| Тригоноцефалия | 20 |
| Асимметрия лица | 30 |
| Гипертелоризм | 45 |
| Аномалии глаз | 75 |
| Колобома (отсутствие фрагмента оболочки глаза) | 15 |
| Аномальное строение носового моста | 80 |
| Большие или неправильно сформированные уши | 85 |
| Патологии строения челюсти | 55 |
| Аномалии строения и работы сердца | 30 |
| Врожденный порок сердца | 20 |
| Аномалии почек | 40 |
| Генитальные аномалии | 45 |
| Анальная атрезия | 7 |
Клинический случай
Ребенок женского пола, родился на 38 неделе беременности, гестационный период осложнен внутриутробной задержкой роста.
Показатели при рождении:
вес — 2400 г;
рост — 42 см;
окружность головы — 31 см.
Данные осмотра:
- отсутствие сосательного рефлекса;
- микроцефалия;
- гипертелоризм;
- катаракта;
- выступающий носовой мостик;
- короткий губной желобок;
- высокое небо;
- короткие пястные кости;
- на левой руке 4 пальца, на правой 5 с гипоплазией большого пальца;
Другие замечания:
- мышечный гипотонус;
- незаращение дужки 3 позвонка;
- гипоплазия мозолистого тела;
- гипоплазия обоих почек;
- дефект межпредсердной и межжелудочковой перегородок, открытие артериального протока;
- маленькие уши неправильной формы;
- неполное прикрепление половых губ;
- отсутствие матки и яичников.
Описан тяжелый случай синдрома Орбели.
Причины
Хромосомы находятся в ядре каждой клетки, несут генетические характеристики. Каждая хромосома имеет короткое плечо и длинное плечо. Далее они подразделяются на полосы (участки, сегменты), которые нумеруются. Например, 22 участок 13 хромосомы.
В случае частичной делеции, то есть удаления сегмента длинного плеча 13 хромосомы обязательно наступают последствия для развития и здоровья ребенка: от малозначительных до глобальных. Понятие синдрома Орбели охватывает делецию проксимальных (центральных) полос длинного плеча 13 хромосомы: с 22 по 31 сегмент. При удалении полос хромосомы в этих пределах наблюдается задержка роста, физические отклонения и умственная неполноценность различной степени тяжести.
Дети с делецией проксимальных участков имеют другие симптомы (не входят в спектр нарушений при синдроме Орбели). Делеция 14 участка создает повышенный риск ретинобластомы (злокачественное новообразование сетчатки). Если затронут 32 сегмент 13 хромосомы, ожидается тяжелая умственная отсталость и другие пороки по аналогии с синдромом Орбели. Делеции 33-34 участка характеризуются изолированными интеллектуальными недостатками и в редких случаях черепно-лицевыми аномалиями, обычно не связаны с другими синдромальными пороками развития. Олигофрения (низкий интеллект) может сопровождаться поведенческими отклонениями или расстройствами аутического спектра.
В большинстве описанных случаев синдрома Орбели исследователи полагают, что делеция спровоцирована нарушениями на начальном этапе эмбрионального развития. В таких случаях родители больного ребенка обычно имеют нормальный кариотип, риск родить второго ребенка с подобной аномалией у них низкий.
Иногда частичная делеция 13 хромосомы спровоцирована хромосомной перестройкой у родителей. Возможна транслокация (смещение и перегруппировка), инверсия (разрыв и присоединение участка в обратном порядке) участков хромосомы. Такие перестановки безвредны для носителя, но могут отразиться на хромосомной организации у потомства.
Диагностика
Диагноз синдрома Орбели можно определить у ребенка пренатально, используя такие тесты:
- УЗИ (нейросонография), пренатальные МРТ и КТ показывают физические аномалии, характерные для генетического расстройства;
- амниоцентез (изучение образца жидкости, взятого из околоплодных вод);
- выбор хорионических ворсинок (исследование образца ткани, взятого из плаценты).
Подтвердить диагноз можно только после рождения с помощью клинического осмотра и анализа крови на хромосомную патологию.
Родители могут сдать анализ на кариотип и пройти генетическое консультирование.
Лечение
Планированием курса терапии и оказанием помощи ребенку занимаются врачи нескольких специальностей: педиатр, невролог, офтальмолог, кардиолог, радиолог, ортопед, хирург, онколог.
В некоторых случаях требуется проведение экстренной хирургической операции для коррекции физических пороков развития. Например, операция может потребоваться, чтобы устранить черепно-лицевые, глазные, генитальные пороки развития и дефекты сердца, связанные с синдромом Орбели или сопутствующими нарушениями, чтобы ребенок мог продолжать жизнедеятельность.
Проблемы со зрением и строением глаз решаются подбором коррекционных линз или очков, хирургической операцией.
При наличии тригоноцефалии или других форм краниостеноза рекомендовано проведение операции по коррекции деформаций свода черепа, причем желательно провести ее в раннем возрасте, пока доступна эндоскопическая хирургия (до 6-10 месяцев). Подробнее об этой аномалии и перспективах читайте в статье о краниостенозе и его формах.
Хирургически исправляется гидроцефалия: показана эндоскопия или шунтирование. Межжелудочковая перегородка корректируется хирургическим способом, но до операции проводят поддерживающее лечение сердечной недостаточности.
Лечение включает регулярный мониторинг неоперабельных нарушений. При ослабленном иммунитете и/или хроническом инфекционном процессе необходимо быстро начинать лечение даже незначительных (как кажется) респираторных заболеваний. Врачи рекомендуют вводить антибиотики перед каждой хирургической операцией, включая удаление зубов (для решения этого вопроса нужна консультация с несколькими специалистами, оценка рисков).
Дальнейшее ведение детей с синдромом Орбели предусматривает симптоматическую и поддерживающую терапию, чтобы помочь им достичь максимального потенциала в физическом и психическом развитии, социализироваться. Такой подход включает коррекционное образование, физиотерапию и другие методики развития.
Прогноз
Рост и взросление при синдроме Орбели осложнены множественными физическими пороками и олигофренией. В младенчестве возможны частые обмороки и судороги. Тщательное наблюдение за пациентом осуществляется, как минимум, до 8 лет.
Дети с тяжелыми пороками развития и развернутой клинической картиной не доживают до 1 года.
Выжившие люди имеют сокращенную продолжительность жизни. Максимальный возраст, зафиксированный у пациента с синдромом Орбели, составил 67 лет.
Большинству взрослых пациентов необходимы вспомогательные услуги для поддержания нормальной жизнедеятельности, включая работу сиделки и домработницы.
Источник
Генетическое строение человека определяется 23 парами хромосом, которые различаются между собой формой и размерами. Самая крупная хромосома 1. Она по размеру превышает хромосому 22 почти в 4 раза. 23 пара – это хромосомы X и Y. Они определяют половую принадлежность людей. У женщин 23 пара содержит 2 хромосомы Х, эта пара обозначается ХХ. У мужчин 23 пара состоит их хромосом Х и Y, т.е. — это пара XY.
Когда в матке будущей матери образуется эмбрион, у него пока нет никаких признаков человека, но 23 пара хромосом уже определяет пол будущего младенца. Предыдущие 22 пары называются аутосомами. Они обеспечивают развитие всех внутренних органов и тканей малыша.
Бесплодие как один из признаков моносомии
Женское бесплодие может быть результатом различных патологий. В случае нарушения строения 23 пары хромосом оно закладывается еще до рождения, когда потенциальная мама сама пребывала в состоянии эмбриона в утробе матери.
Если в паре ХХ отсутствует одна хромосома Х или она повреждена, такая патология называется моносомией Х. Бесплодие – один из наиболее показательных признаков моносомии Х. Современная медицина изучает как признаки этой патологии, так и способы ее лечения.
Что это такое?
Моносомия — это отклонение, названое по имени открывшего его ученого синдромом Тернера. Эта генетическая болезнь бывает только у девочек. По данным статистики ее обнаруживают примерно в 1 из 2500 случаев.
На данный момент развития медицины предотвратить синдром Тернера невозможно. Девочки с этой патологией могут вести нормальную жизнь, но им необходимо наблюдение врача, поскольку могут развиться осложнения.
Другие признаки моносомии
Выявлено, что у девочек с вероятностью иметь синдром моносомии еще в детском возрасте могут быть следующие признаки болезни:
- В младенческом возрасте отекают ручки и стопы ног.
- Малышки отстают от сверстниц по росту.
- Нёбо у таких девочек расположено выше, чем у среднего ребенка, а ушки ниже.
- Больные девочки склонны к набиранию излишнего веса.
- Часто наблюдается плоскостопие.
- Веки нависают низко над глазами.
- Косоглазие.
- Шея укорочена.
- Наблюдается оволосение частей тела.
- Укорочены кисти рук.
Кроме перечисленных болезней, у подростка-девочки могут быть выявлены:
- Порок сердца (несмыкаемость стенок клапанов).
- Замедление наступления половой зрелости.
- Проблемы со слухом.
- Частое повышение артериального давления (гипертония).
- Постоянные болезни, связанные с инфекциями в ушах.
- Сухость в глазах.
- Нарушение осанки, вплоть до сколиоза.
С точки зрения полового развития наблюдается:
- Непропорциональность наружных половых органов.
- Неразвитость молочных желез.
- Отсутствие оволосения подмышками и над лобком.
- Недоразвитость матки.
- Отсутствие или пассивность яичников.
- Пониженное содержание женских гормонов.
- Менструальная пассивность, вплоть до отсутствия месячных.
Трудность постановки диагноза по симптомам состоит в том, что такие же признаки могут наблюдаться при других болезнях. Если они есть у девочки любого возраста, необходимо пройти обследование в поликлинике.
Одним из характерных признаков моносомии по половым хромосомам является избыток кожи на боковых сторонах шеи, который образует продольные складки.
Причины появления моносомии
На данном этапе развития науки детально установить причины возникновения у плода моносомии не удалось. То, что известно наверняка – патология не связана с наследственностью. Основное влияние оказывают на возникновение патологии у плода генные нарушения половых клеток отца в период зачатия. В этом главные причины моносомии.
Если гены отца претерпели мутацию, произошла перестройка структуры Х-хромосомы, у ребенка — девочки тоже может быть повреждена 23 пара хромосом. Но надо отметить, что в такой семье ставить крест на рождении детей не стоит. У отца с нарушением Х-хромосомы очень вероятно зачатие и рождение вполне здорового ребенка.
Почему появляется моносомия Х-хромосомы? Причины данной болезни изучаются, но на данном этапе медицина может сказать следующее:

- Возникновение у плода моносомии не связано с неблагоприятными внешними условиями.
- Эта патология не передается по наследству. Иначе ее получали бы все девочки в семье генно-травмированного отца.
- Наиболее вероятная причина синдрома Тернера – влияние вредных факторов на половые клетки отца незадолго до зачатия.
- В период деления зиготы и превращения ее в эмбрион могут происходить сбои, являющиеся причиной мозаичного типа моносомии.
Диагностика заболевания
Чтобы моносомия хромосомы у плода была исключена, еще до родов находящимся в группе риска женщинам делают специальное генетическое обследование. Это обследование — пренатальный генетический анализ – включает в себя ультразвуковое обследование, биопсию мышц и кожи плода, лабораторный анализ крови.
Такие серьезные обследования врач назначает, когда речь идет о том, оставить плод или прервать беременность. Поэтому их назначают в первые 3 месяца беременности. То есть, если семья не решает вопрос, быть или не быть ребенку, обследования не назначают.
Врач может рекомендовать генетическое обследование, чтобы моносомия по 21 хромосоме была исключена, если:
- Будущей матери больше 35 лет.
- В семье беременной наблюдались наследственные патологии.
- В семье наблюдались случаи ломких (фрагильных) Х хромосом.
- Подозрение на гемоглобинопатию.
- Подозрение на синдром Дауна у ребенка и другие, связанные с наследственностью, неблагоприятные ситуации.
При диагностике учитываются такие моменты, что обследование не должно приносить вред здоровью матери или ребенка, оно не может привести к потере плода. Врачи, проводящие диагностику, обязаны иметь высокую квалификацию для постановки диагноза по результатам обследования.
До проведения генетического обследования женщина должна пройти скрининг-обследование у гинеколога в своей поликлинике, чтобы врач принял решение, относится ли беременная к группе риска, т. е., есть ли вероятность, что у плода выявится синдром моносомии. Только после положительного ответа на этот вопрос беременная направляется на генетическое обследование.

В пренатальной диагностике существуют и другие методы – прокол плодного пузыря с целью взятия на анализ околоплодной жидкости, взятие крови из пуповины, фетоскопия. Последняя представляет собой введение зонда и осмотр плода через него. Фетоскопия используется очень редко, поскольку на современном уровне развития медицинской аппаратуры эта процедура с успехом заменяется УЗИ.
Если у плода не была вовремя обнаружена моносомия по Х-хромосоме, а появилось подозрение на диагноз через несколько лет после рождения, проводится обследование девочки следующими способами:
- Определяется отсутствие полового хроматина.
- Определяется кариотип.
- Проводится консультация детского гинеколога.
- Делают УЗИ внутренних половых органов.
- В возрасте 9-10 лет проводится анализ крови на гормоны.
- Назначается рентген кистей на соответствие костного развития возрастной норме.
По результатам обследования специальной комиссией может быть принято решение об инвалидности.
Типы моносомии

Моносомия — это болезнь, которая бывает разных типов. У рожденной девочки вторая хромосома в паре ХХ может отсутствовать совсем. Такой тип генетики обозначают как 45Х0. У этих девочек полностью отсутствуют или неразвиты яичники и матка. Их половое созревание не состоится. Отсутствует хромосома, которая определяет развитие вторичных половых признаков, самих половых органов и соответствующих гормонов.
Если вторая хромосома в паре ХХ присутствует, генетический тип 46 XY. В данном случае девочка будет обладать маткой и яичниками, но они будут иметь рудиментарное строение, т. е. она не будет способна к овуляции и зачатию ребенка.
Гораздо более опасен случай, когда одной хромосомы не хватает в парах 1 – 22. Их называют аутосомами. При нормальном развитии зиготы в каждой клетке 2 хромосомы направляются к разным ее полюсам. Если одной аутосомы не хватает, к полюсу клетки направляется только существующая хромосома. При ее слиянии со здоровой, зигота получается с аномальным количеством хромосом. Это приводит к гибели зародыша и самопроизвольному прерыванию беременности. Если число хромосом больше на 1, чем в стандартном варианте, выкидыша может и не произойти, но тогда рождается ребенок, неспособный долго жить.
Частичные моносомии
Частичная моносомия образуется при поломке хромосомы, вследствие чего в паре остается одна целая и часть второй хромосомы. Причиной патологии является структурная хромосомная перестройка половых клеток родителей в период незадолго до зачатия. Последствия заболевания зависят от того, какая именно хромосома подверглась частичному разрушению.
Синдром «кошачьего крика»
Разрушение части хромосомы в 5 паре создает синдром «кошачьего крика». Гортань у больного ребенка сужена, хрящи мягкие, в результате плач младенца напоминает кошачье мяуканье.
Для этого типа характерна также мышечная вялость малыша, лунообразное личико, умственное недоразвитие и физическое отставание по сравнению со своими сверстниками. У ребенка наблюдается недоразвитие черепа, низкое расположение ушей, косоглазие и проблемы со зрением. Как правило, такие дети страдают пороками сердца. Живут эти больные недолго – срок достигает не более 10 лет. Подобная патология замечается в 1 из 45 000 случаев.
Синдром Вольфа-Хиршхорна
Разрыв одной хромосомы из 4 пары (синдром Вольфа-Хиршхорна) возникает в 1 случае из 100 000. Дети, у которых моносомия хромосомы из 4 пары, чаще получаются у молодых мамы и папы. Вес ребенка при рождении значительно меньше нормы – примерно 2 кг. У младенцев наблюдается задержка физического и психического развития, моторика усложняется очень медленно. Они страдают недоразвитием черепа.
По внешним признакам можно отметить клювовидный нос, выступающий лоб, низкую посадку ушей, продольные складки перед ушами. У них тоже присутствует мышечная вялость. Они почти не реагируют на внешние раздражители. Такие дети подвержены судорогам. Внешне у них могут быть аномальное строение глаз, маленький рот с опущенными губами в уголках, искривление стоп. Больные дети часто страдают пороками сердца. Подвержены заболеваниям почек. Большая часть детей с этим хромосомным разрушением не доживают до года.
Синдром Орбели
Разрыв хромосомы из 13 пары называют синдромом Орбели. Дети с этим отклонением рождаются с малым весом – до 2,2 кг. Имеют патологии практически всех внутренних органов. У них недоразвит череп, отсутствует переносица – лоб сразу переходит в нос. Нос имеет увеличенную ширину, небо располагается высоко, ушные раковины занижены. У младенца наблюдаются патологии зрения, недоразвитие опорно-двигательного аппарата. Может отсутствовать естественный канал прямой кишки и задний проход. Часто наблюдаются пороки сердца, болезни почек, патологии головного мозга. Эти дети, как правило, олигофрены, их мучают судороги, они часто теряют сознание. Младенцы не доживают до 1 года.
Моносомия — это весьма опасное заболевание, при этом детки с частичной моносомией рождаются крайне редко. Будущим мамам не надо настраиваться на столь печальное событие.
Осложнения моносомии Х
Женщины с моносомией Х подвержены различным заболеваниям внутренних органов чаще, чем генетически здоровые люди. Они должны наблюдаться у врача и периодически проходить диспансеризацию.
Часто встречающимся осложнением является патология почек. У больных женщин нередко возникают инфекционные болезни мочеполовых органов, неправильно расположены, не полностью развиты почки.
Если моносомия у человека имеет место быть, то иногда страдает щитовидная железа, в которой вырабатывается недостаточный уровень гормонов, необходимых организму. Щитовидная железа воспаляется, из-за чего и происходят эти неприятности. Лечением может служить только прием препаратов, поддерживающих нормальный гормональный фон. Их должен назначить врач. Самолечение недопустимо.
Страдающие синдромом Тернера подвержены заболеванию целиакии. Эта патология выражается в возникновении аллергии на белок глютен. Больным нельзя есть изделия из пшеницы и ячменя.
Осложнения может давать любая моносомия. Болезни, которые сопровождают моносомию, могут быть разными, например, сердечно-сосудистые заболевания. Женщинам с патологией необходимо наблюдение кардиолога, который проследит за состоянием их аорты и не допустит развития гипертонии.
Поскольку больные синдромом Тернера склонны к ожирению, им может грозить сахарный диабет.
Замершая беременность
Генетические патологии, в том числе моносомию плода, считают одной из причин развития замершей беременности. Это гибель плода внутри утробы матери. При моносомии это случается до 12 недели беременности. Иногда срок жизни плода больше – до 20 недель. Такая опасность грозит при отсутствии у плода одной из хромосом из пар 1–22, это так называемая трисомия. Моносомия является поводом для осознанного прерывания беременности.
Пытаться сохранить плод с серьезными аномалиями развития не представляется осмысленным. Замершая беременность заканчивается выкидышем. Такому риску подвержены женщины в возрасте старше 35 – 40 лет, а также те, у кого это случалось ранее.
Учеными также отмечено уменьшение продолжительности жизни женщин с моносомией, склонность к атеросклерозу, опухолям.
Лечение моносомии

Моносомия — это патология, которая лечению не поддается, но врачи успешно справляются с ее последствиями. Суть лечения состоит в том, что лишенный гормонального регулирования яичниками организм нуждается в искусственном регулировании. Патологию внутренних органов лечат медикаментозно или хирургическим путем. Комплексное лечение включает:
- Прием препаратов гормонов роста девочками сразу после постановки диагноза.
- К возрасту полового созревания здоровых детей, страдающим моносомией, выписывают половые гормоны.
- Желающим выносить ребенка женщинам с патологией помогает репродуктивная медицина – внесение донорской оплодотворенной яйцеклетки с дальнейшим наблюдением за протеканием беременности.
- Лечение патологий внутренних органов современными препаратами и процедурами.
- Психологическая помощь женщинам с целью их адаптации в социальной среде с учетом особенностей физиологии.
Наиболее важным моментом в лечении является своевременное его начало – сразу после постановки диагноза. Второй момент – учет вида моносомии. Если ее тип 45Х0, то женщина не сможет иметь своего ребенка никакими способами. Психологическая помощь должна подготовить ее к мысли об усыновлении ребенка — сироты.
Мозаичный вид моносомии Х — 46XY при современном развитии медицины дает возможность выносить своего ребенка. Но он может родиться больным. Усыновление является более предпочтительным и в этом варианте.
Источник