Синдром недержания пигмента у детей


Синдром Блоха-Сульцбергера – наследственная форма нарушения пигментации кожи, которая часто сочетается с пороками развития зубов, волос, ногтей и глаз. Симптомы заболевания характеризуются выраженной стадийностью – сначала на коже появляется эритематозная сыпь в виде пятен и линий, затем на ее месте развивается гиперкератоз, сменяющийся пятнами и последующей гипопигментацией с атрофией кожных покровов. Диагностика синдрома Блоха-Сульцбергера производится на основании данных настоящего статуса больного, гистологического исследования образцов кожи в области поражения, изучения наследственного анамнеза и молекулярно-генетических анализов. Специфического лечения этой патологии на сегодняшний момент не существует, используют симптоматические и поддерживающие мероприятия различного характера.
Общие сведения
Синдром Блоха-Сульцбергера (семейная форма недержания пигмента, нейрокожный меланобластоз) – генетическое заболевание, характеризующееся нарушением метаболизма меланина в коже и рядом сопутствующих пороков развития. Впервые это заболевание было описано в 1926 году швейцарским дерматологом Б. Блохом, затем более детальное изучение данной патологии провел американский педиатр М. Сульцбергер в 1929 году и, независимо от предыдущих исследователей, немецкий врач Г. Сименс. Именно поэтому в литературе можно найти другое название этого заболевания – синдром Блоха-Сименса. Удалось выяснить, что патология наследуется сцепленно с Х-хромосомой, при этом мутантный аллель является доминантным. По этой причине синдром Блоха-Сульцбергера во много раз чаще встречается у девочек – половое распределение составляет примерно 6:210, так как наличие этой мутации у эмбрионов мужского пола практически всегда является летальным и приводит к самопроизвольному прерыванию беременности. Развитие заболевания у мальчиков может быть обусловлено генетическим мозаицизмом, наличием сопутствующего синдрома Клайнфельтера или редких точечных «мягких» мутаций. Общая встречаемость синдрома Блоха-Сульцбергера составляет примерно 1 случай на 75 000 новорожденных.

Синдром Блоха-Сульцбергера
Причины синдрома Блоха-Сульцбергера
При синдроме Блоха-Сульцбергера происходит повреждение гена IKBKG, который располагается на Х-хромосоме. Продуктом его экспрессии является многофункциональный сложный белок – регуляторная субъединица NEMO-ингибиторной киназы, участвующей в сигнальной системе важного транскрипционного фактора (NF-каппа-B). Этот фактор и соответствующий ему сигнальный путь регулирует огромное количество различных процессов в организме человека – участвует в процессах адаптации при стрессе, иммунном ответе, некоторых формах воспалительных реакций, процессах клеточной адгезии, а также тормозит процессы апоптоза. Наиболее часто причиной синдрома Блоха-Сульцбергера становятся крупные транслокации и делеции гена IKBKG, в результате чего экспрессия и выделение белка с этого гена полностью прекращаются.
Поскольку у женщин имеется две Х-хромосомы, при наличии второй нормальной аллели гена IKBKG такая мутация не угрожает жизни, но обуславливает развитие синдрома Блоха-Сульцбергера. Известен факт, что в соматических клетках женского организма всегда активна только одна Х-хромосома, тогда как вторая сконденсирована в половой хроматин. Значительная вариабельность выраженности симптомов заболевания обусловлена распределением клеток, где активна хромосома именно с мутантной формой гена IKBKG. Как следствие, в вышеуказанных клетках не образуется регуляторной субъединицы NEMO-ингибиторной киназы, что и приводит к характерным порокам развития, формирующим клиническую картину синдрома Блоха-Сульцбергера. Кожные симптомы связаны с нарушением проницаемости мембран меланоцитов (в результате чего практически весь пигмент беспрепятственно покидает клетки) и аутоиммунными реакциями.
В отличие от женщин, у мужчин в норме есть лишь одна Х-хромосома, поэтому при наличии нонсенс-мутации в гене IKBKG выделение важного белка не происходит абсолютно во всех клетках организма. Это становится причиной массированного апоптоза гепатоцитов еще на этапе внутриутробного развития – в норме этот процесс задерживается как раз системой NF-каппа-B. Развитие нарушений, подобных синдрому Блоха-Сульцбергера, у мальчиков возможно при наличии сопутствующего синдрома Клайнфельтера (кариотипа XXY) или генетического мозаицизма, когда только часть клеток в организме имеет дефект гена IKBKG. В последние годы были выявлены точечные мутации этого гена, которые не приводят к полной остановке транскрипции, но изменяют структуру конечного белка. Однако чаще всего у мальчиков с такими дефектами возникает не синдром Блоха-Сульцбергера, а другие генетические заболевания – эктодермальные дисплазии, иммунодефициты, пороки развития скелета.
Симптомы синдрома Блоха-Сульцбергера
Одним из наиболее выраженных и распространенных проявлений синдрома Блоха-Сульцбергера является дерматоз, который обнаруживается при рождении или (реже) возникает на протяжении первых дней жизни новорожденного. В развитии изменений кожных покровов при этой патологии наблюдается характерная стадийность, что также является важным диагностическим признаком. Локализация таких изменений – на боковых поверхностях конечностей, туловища, шеи, вдоль линий Шарко или проекций основных нервных стволов. В большинстве случаев выделяется четыре основных стадии кожных симптомов синдрома Блоха-Сульцбергера:
1-я стадия – воспалительная или везикулобуллезная. Начинается при рождении больного или на протяжении 2-3 недель жизни и длится до возраста 3-8 месяцев. На этом этапе заболевания возникают везикулы, эритематозная сыпь, возможно развитие пузырей и пустул. С учетом возраста больных синдромом Блоха-Сульцбергера существует определенный риск инфекционных осложнений на пораженных участках кожи.
2-я стадия – гипертрофическая или веррукозная. Характеризуется развитием на пораженных участках тела гиперкератоза в виде бляшек, бородавчатых и лихеноидных разрастаний. Их распределение, как правило, симметричное и линейное, вдоль линий Шарко или проекций нервных стволов. Длительность этой стадии синдрома Блоха-Сульцбергера составляет несколько месяцев (до возраста одного года), у некоторых больных может отсутствовать.
3-я стадия – пигментная. На этом этапе заболевания у больных на пораженных участках кожи возникают очаги гиперпигментации различных форм и размеров темно-коричневого цвета. Почти у половины пациентов с синдромом Блоха-Сульцбергера такие очаги появляются на неизмененных участках тела и не связаны с высыпаниями, характерными для предыдущих стадий. Длительность гиперпигментации составляет несколько лет, обычно – до периода полового созревания.
4-я стадия – атрофическая. Характеризуется потерей пигмента на очагах поражения с развитием признаков атрофии кожи. У некоторых больных синдромом Блоха-Сульцбергера такие проявления могут быть выражены очень слабо, в отдельных случаях симптомы заболевания полностью исчезают после завершения полового созревания.
Помимо кожных проявлений, синдром Блоха-Сульцбергера может приводить к развитию очаговой алопеции, дистрофии ногтей. Почти у 80% больных отмечаются аномалии зубного ряда – искривления, отсутствие зубов. В половине случаев заболевания выявляются нарушения зрения – катаракта, косоглазие, атрофия зрительного нерва и некоторые другие расстройства. Умственное развитие при синдроме Блоха-Сульцбергера обычно не страдает, но возможна некоторая задержка. В редких случаях отмечается олигофрения. Все проявления заболевания имеют тенденцию к ослаблению после завершения подросткового периода.
Диагностика и лечение синдрома Блоха-Сульцбергера
Для определения синдрома Блоха-Сульцбергера используют множество диагностических методов и техник – дерматологический осмотр, изучение наследственного анамнеза, гистологическое исследование пораженных участков кожных покровов, молекулярно-генетические анализы. При осмотре выявляются разнообразные (в зависимости от возраста больных и стадии заболевания) изменения кожи эритематозного, везикулярного или гиперкератического характера, у старших пациентов может определяться очаговая гипер- или гипопигментация кожи. Помимо этих проявлений, при синдроме Блоха-Сульцбергера возможна дистрофия ногтей, алопеция, аномалии строения зубов.
Наследственный анамнез может выявить семейный, доминантный и сцепленный с Х-хромосомой характер наследования патологии. В некоторых случаях у матери больной в анамнезе отмечается несколько случаев самопроизвольного прерывания беременности – это связано с внутриутробной смертью плода мужского пола. Результаты гистологического исследования тканей кожи при синдроме Блоха-Сульцбергера зависят от стадии заболевания – на первом этапе обнаруживается спонгиоз, развитие эпидермальных пузырей, заполненных эозинофилами и фибриновыми массами. На второй стадии патологии выявляются признаки внутриэпителиальной кератинизации, акантоз и гиперкератоз, в дерме отмечается отек с нейтрофильной и эозинофильной инфильтрацией. На третьей стадии синдрома Блоха-Сульцбергера воспалительные изменения в дерме (отек, инфильтрация) исчезают, но наблюдается значительное накопление пигмента в верхних слоях кожи. Четвертая стадия характеризуется исчезновением пигмента, развитием фиброзной ткани и частичным исчезновением придатков кожи.
Молекулярно-генетическая диагностика синдрома Блоха-Сульцбергера выполняется врачом-генетиком и может быть произведена несколькими основными техниками. Прямое автоматическое секвенирование последовательности гена IKBKG позволяет выявить практически любые изменения в его структуре. Транслокации и делеции значительных участков гена, часто выступающие в качестве причины синдрома Блоха-Сульцбергера, можно обнаружить при помощи методики FISH-анализа. Данное заболевание также может быть подтверждено посредством исследования инактивации Х-хромосом в клетках пораженных тканей.
Специфического лечения синдрома Блоха-Сульцбергера на сегодняшний момент не существует, кожные поражения на воспалительном этапе заболевания обрабатываются антисептическими средствами и растворами для предотвращения инфекционных осложнений. Кроме того, рекомендуется местное назначение глюкокортикоидных стероидов для уменьшения воспаления, однако такое лечение следует производить с осторожностью, учитывая высокую проницаемость кожных покровов у детей младшего возраста. Другие проявления синдрома Блоха-Сульцбергера (пороки развития зубов, глаз) лечат при наличии показаний.
Прогноз и профилактика синдрома Блоха-Сульцбергера
Прогноз синдрома Блоха-Сульцбергера чаще всего благоприятный, так как с момента начала полового созревания проявления заболевания значительно ослабевают. Ухудшить прогноз могут изменения внутренних органов, нервной системы, глаз, нарушения которых изредка наблюдаются при этой патологии. Кроме того, в отдельных случаях синдрома Блоха-Сульцбергера может возникать первичный иммунодефицит, который также значительно ухудшает перспективы заболевания. Профилактика патологии сводится только к пренатальной диагностике и медико-генетическому консультированию родителей при отягощенной наследственности у будущей матери.
Источник
Недержание пигмента у новорожденного
Недержание пигмента — нейрокожный синдром, названный так за особенные «мраморные» разводы коричневого пигмента, которые появляются на туловище и иногда на конечностях в позднем грудном и раннем детском возрасте.
Недержание пигмента — наследственное, мультисистемное заболевание, для которого характерна ретикулярная гиперпигментация по типу «размазанных» пятен. У 90% пораженных новорожденных детей изменениям в пигментации предшествует фаза воспаления.
У новорожденных пятна эритемы и пузыри рассеяны на туловище, волосистой части головы и на конечностях. Пузыри типично ориентированы по линиям Блашко, особым эмбриональным разделительным линиям. Хотя воспалительные очаги могут рецидивировать месяцами, они обычно сменяются гиперкератотическими бородавчатыми бляшками в возрасте от нескольких недель до нескольких месяцев.
После гиперкератотической стадии, в возрасте 2-6 мес, типично развивается усиленная пигментация. В позднем детском возрасте коричневые полоски часто исчезают, оставляя атрофические пятна гипопигментации. Хотя эти незаметные рубцы могут быть единственным проявлением кожного заболевания у детей старшего возраста и у взрослых, отсутствие зубов, пороки зубочелюстной системы и, редко, дистрофия ногтей в сочетании с рецидивирующими или стойкими подгнотевыми бородавчатыми узелками могут дать дополнительные ключи к диагнозу.
Гистопатологическое исследование пузырей у новорожденных выявляет характерный внутриэпидермальный отек и везикулы, наполненные эозинофилами. При подозрении на недержание пигмента ребенка необходимо обследовать на сочетанные дефекты, которые могут поражать ЦНС, сердце, глаза, скелетную и зубочелюстную системы.
Недержание пигмента наследуется как Х-сцепленное доминантное заболевание, летальное для мужского пола. 97% пораженных детей — девочки. Тщательное обследование матерей часто выявляет незаметные кожные признаки. Мутация в гене NEMO идентифицирована у 85% обследованных пациентов.
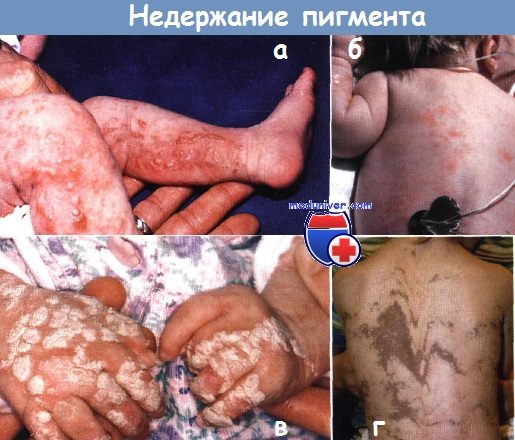
Недержание пигмента.
Везикулы на эритематозном основании расположены линейно на (а) ногах и (б) туловище 8-дневной девочки
в — бородавчатые папулы развились на пальцах этой 4-месячной девочки на месте везикул
г — прогрессирующая гиперпигментация по типу «мраморных» разводов впервые замечена на туловище этой пациентки в 4-месячном возрасте
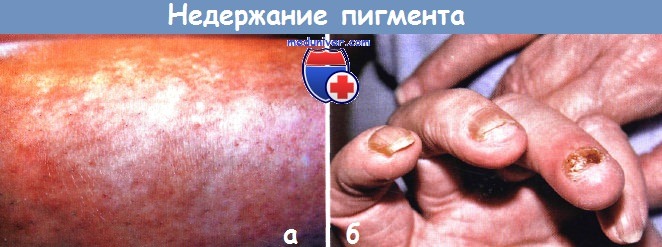
Недержание пигмента:
а — незаметное рубцевание на ногах было единственным признаком недержания пигмента. Обратите внимание на отсутствие фолликулов в линейных рубцах
б — у этой 60-летней женщины с недержанием пигмента рецидивирующие бородавчатые подногтевые узелки и дистрофия ногтей наблюдались с подросткового возраста.
Биопсия показала эозинофильное воспаление дермы и дискератоз.
— Также рекомендуем «Сосудистые аномали (пороки развития) у новорожденного»
Оглавление темы «Пятна кожи новорожденных»:
- Мастоцитоз у новорожденного
- Недержание пигмента у новорожденного
- Сосудистые аномали (пороки развития) у новорожденного
- Пятна лососевого цвета у новорожденного
- Винные пятна у новорожденного
- Врожденная мраморная телеангиэктатическая кожа (CMTC) у новорожденного
- Венозная мальформация у новорожденного
- Лимфатическая мальформация у новорожденного
- Артериовенозные мальформации у новорожденного
- Гемангиома у новорожденного
Источник
Определение
Недержание пигмента, или синдром Блоха-Сульцбергора (Block-Sukberger syndrome) это редкий синдром с доминантным Х-сцепленным типом наследования, который включает глазные, кожные, скелетные и другие системные аномалии, а также изменения центральной нервной системы.
Патогенез заболевания неизвестен. Это доминантное, сцепленное с Х-хромосомой заболевание, которое обычно приводит к летальному исходу у мужчин и поэтому наблюдается только у младенцев женского пола. Однако ребёнок мужского пола с недержанием пигмента и одновременно с синдромом Клайнфелтера (Klinefelter syndrome) (XXY) или генетической мозаичностью может выжить.
Анамнез
Характерные кожные изменения обычно появляются через несколько дней после рождения, а глазные изменения в младенчестве или более старшем возрасте.
Важные клинические признаки
Приблизительно у 1/3 всех детей с недержанием пигмента развиваются глазные проявления. Изменения на глазном дне включают:
• расширенные извитые сосуды сетчатки;
• отсутствие капиллярной перфузии на периферии с артериовенозными анастомозами и неоваскуляризацией (рис. 8-11, 8-12);
• гипоплазию центральной ямки;
• окклюзию артериальных ветвей;
• неоваскуляризацию диска зрительного нерва;
• стягивание сетчатки;
• тракционную и регматогенную отслойку сетчатки;
• ретинальные складки;
• витреальные кровоизлияния;
• побледнение диска зрительного нерва.
.")
Рис. 8-11. Недержание пигмента. Периферия глазного дна пациента с недержанием пигмента, определяют отсутствие периферической капиллярной перфузии сетчатки и артериовенозные анастомозы (стрелка).
.")
Рис. 8-12. Недержание пигмента. На флюоресцентной ангиограмме глазного дна пациента с недержанием пигмента на периферии определяются отсутствие периферической капиллярной перфузии сетчатки и артериовенозные анастомозы (стрелка).
Глазные аномалии нередко крайне асимметричны.
Сопутствующие клинические признаки
Другие глазные признаки включают катаракту, конъюнктивальную пигментацию и косоглазие. Кожные изменения, из-за которых заболевание получило своё название, заключаются в появлении везикулярной кожной сыпи (рис. 8-13), которая позже трансформируется в демигментированные участки (рис. 8-14). Эти изменения начинают развиваться через несколько дней после рождения ребёнка.
Зубные аномалии включают потерю зубов или развитие зубов конусообразной формы, что наблюдают примерно у 2/3 пациентов (рис. 8-15). Сопутствующие аномалии центральной нервной системы включают судороги, спастический паралич и умственную отсталость.
Дифференциальная диагностика
Если заболевание выявляется на стадии отслойки сетчатки, необходимо учитывать другие причины разтированные участки (рис. 8-14). Эти изменения начинают развиваться через несколько дней после рождения ребёнка.
Зубные аномалии включают потерю зубов или развитие зубов конусообразной формы, что наблюдают примерно у 2/3 пациентов (рис. 8-15). Сопутствующие аномалии центральной нервной системы включают судороги, спастический паралич и умственную отсталость.

Рис. 8-13. Недержание пигмента, дерматологические признаки. Везикулярные патологические изменения кожи у ребёнка с недержанием пигмента.
Рис. 8-14. Недержание пигмента, дерматологические признаки. Пигментные изменения кожи у пациента с регрессировавшей везикулярной сыпью при недержании пигмента.

Рис. 8-15. Недержание пигмента, изменения зубов. Рентгенограмма зубов с коническими зубами у пациента с недержанием пигмента.
Дифференциальная диагностика
Если заболевание выявляется на стадии отслойки сетчатки, необходимо учитывать другие причины развития отслойки сетчатки в младенческом возрасте (см. табл. 8-3).
Диагностика
Необходимо клиническое обследование, включающее исследование кожи.
Прогноз и лечение
Большинству пациентов с недержанием пигмента лечение не требуется. Прогрессирующая периферическая неоваскуляризация сетчатки отвечает на лечение лазерной фотокоагуляцией или на криотерапию. При отслойке сетчатки выполняют склеральное пломбирование и/или витрэктомию.
С.Э. Аветисова, В.К. Сургуча
Опубликовал Константин Моканов
Источник