Синдром нагера или акрофациальный дизостоз нагера

Среди генетических заболеваний одним из самых редких признана патология, которую впервые в середине прошлого века описали учёные из Швейцарии Рейнер и Нагер. В честь второго заболевание и было названо. С тех пор синдром Нагера постоянно изучают врачи всех стран, стараясь найти способ предотвратить появление болезни или помочь больному жить, не страдая от собственной неполноценности. Сегодня врачи способны поставить точный диагноз, выявив болезнь на ранних сроках беременности. С помощью ультразвукового исследования медики получают достоверную информацию о состоянии костей и слухового прохода плода, находящегося в утробе матери.
Особенности недуга
Заболевание, которое описали учёные в конце 40-х годов ХX столетия, встречается редко, но каждый отдельный случай заслуживает внимания медиков. Давно доказано, что синдром Нагера – результат генной мутации, справиться с которой почти невозможно, но врачи обнаруживают признаки патологии при первом посещении беременной женщиной кабинета УЗИ.
С помощью ультразвука обнаруживают:
- Недостаточный уровень развития костей нижней челюсти плода или полное отсутствие челюстных сочленений.
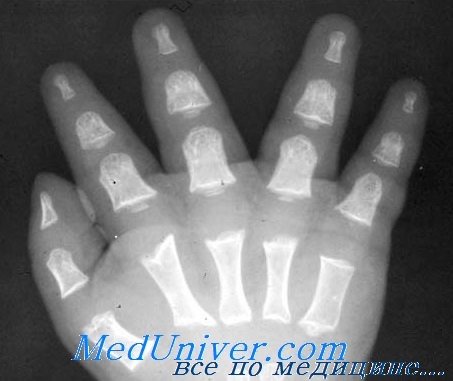
- Недоразвитие или отсутствие костей, идущих от запястья к пальцам.
- Изменения в формировании костей пальцев рук (6 и более пальцев или их отсутствие).
- Неправильную (аномальную) форму разреза глаз (опущение верхнего или нижнего века).
- Нарушения строения и формирования слухового аппарата.
Если у ребёнка, появившегося на свет, сразу после рождения обнаруживается болезнь, которую описал Нагер, значит, малыш будет абсолютно глухим, а челюстная аномалия станет причиной нарушенной артикуляции. Однако у детей с таким диагнозом нормальный интеллект, несмотря на задержку в психомоторном развитии.
Количество отклонений от нормы в формировании различных органов и систем детского организма не одинаково у всех пациентов.
Проявления патологии проявляются в виде аномальных отклонений от нормы:
- Гидроцефалия и сколиоз.
- Микроцефалия и недоразвитие шейного отдела позвоночника.
- Нарушение в развитии полушарий головного мозга.
- Стеноз ликворного русла.
- Порок сердца и аномальное строение кровеносных сосудов.
Встречаются случаи, когда малыш появляется на свет с пороками развития не совместимыми с жизнью, но в основном новорождённые выживают и живут на протяжении долгого времени.
Причины возникновения патологии и её симптоматика
В чём причина генетического сбоя, учёные не знают и сегодня. Однако доподлинно известно, что с синдромом, который описал Nager, рождаются дети не у всех родителей, имеющих мутировавший ген.Порой даже если отец и мать носители такого гена, их малыш может родиться абсолютно здоровым, а у здоровых мужчин и женщин рождаются больные дети. Причина – спонтанная мутация:
- Это значит, что будущая мать подверглась радиоактивному облучению во время беременности или жила в населённом пункте, расположенном неподалёку от места разработки и добычи радиоактивных руд.
- Не менее опасно для будущих младенцев увлечение их родителей наркотическими веществами или алкоголем.
- Токсикомания приводит к видоизменению генов и спонтанной мутации. Результат – рождение малыша с синдромом Нагера.
 Как утверждают учёные-медики, синдром Нагера диагностируется у половины детей, получивших мутировавший ген от обоих родителей. Вторая половина таких новорождённых – абсолютно здоровые дети.
Как утверждают учёные-медики, синдром Нагера диагностируется у половины детей, получивших мутировавший ген от обоих родителей. Вторая половина таких новорождённых – абсолютно здоровые дети.
Риск произвести на свет больного малыша слишком велик, поэтому все женщины, ставшие на учёт в женской консультации, стремятся пройти обследование и получить ответ на свой вопрос.
Результаты УЗИ позволят принять правильное решение, спланировать ведение беременности и принять решение относительно способа родоразрешения.
Характерный симптом акрофациального дизостоза (синдрома Нагера) – аномалии в развитии нижней челюсти. Этот челюстно-лицевой дизостоз сочетается с другими признаками заболевания:
- Недоразвитие или отсутствие луча первого пальца кистей рук (мизинцев). В некоторых случаях отсутствует или недостаточно развит луч безымянного пальчика.
- Недостаточное развитие лучевых костей и, соответственно, неестественное положение пальцев рук.
- Укорочение твёрдого нёба.
- Недоразвитие коренных зубов.
- Неправильная форма ушных раковин.
- Антимонголоидный разрез глаз (опущение верхнего или нижнего века).
У детей, рождённых с синдромом Нагера, могут быть искривлены нижние конечности или проявляется дизостоз на других участках тела (недоразвиты и деформированы ключицы, лопатки, скулы).
Чего ждать в будущем
 Прогноз, о котором говорят врачи, не столь печален. Существуют определённые методы лечения, применяемые при наличии незначительных аномалий в развитии ребёнка. В большинстве случаев проводится операция по устранению деформаций, в ходе которой:
Прогноз, о котором говорят врачи, не столь печален. Существуют определённые методы лечения, применяемые при наличии незначительных аномалий в развитии ребёнка. В большинстве случаев проводится операция по устранению деформаций, в ходе которой:
- Восстанавливается форма кистей и пальцев рук (разделение сросшихся фаланг).
- Коррекция формы ушных раковин.
- Восстановление внутренних органов.
Если диагноз был поставлен на ранних сроках после зачатия, родителям предлагают согласиться на искусственное прерывание беременности (аборт).
Существуют и более сложные техники хирургического вмешательства, применяемые очень редко. Современные акушеры-гинекологи в некоторых случаях с помощью кесарева сечения частично извлекают плод из полости матки, проводят трахеотомию, обеспечивая крохе возможность самостоятельно дышать, затем операция завершается.
В других случаях малышу хирургическим путём формируют слуховой проход или вживляют специальный аппарат. С его помощью ребёнок слышит. Это вмешательство называется бинауральное слухопротезирование. Во время операции врачи используют имплантируемые цифровые слуховые аппараты, обеспечивающие качественное восприятие звуков.
 Большую работу проводят стоматологи и хирурги, проводящие челюстно-лицевые операции. Их цель – восстановление формы нижней и верхней челюсти, зубного ряда. С помощью данного вмешательства маленькому пациенту обеспечивают возможность не только разговаривать, но и пережёвывать пищу. Деятельность ортопедов направлена на устранение деформаций верхних и нижних конечностей, восстановление их функциональности и качественной подвижности.
Большую работу проводят стоматологи и хирурги, проводящие челюстно-лицевые операции. Их цель – восстановление формы нижней и верхней челюсти, зубного ряда. С помощью данного вмешательства маленькому пациенту обеспечивают возможность не только разговаривать, но и пережёвывать пищу. Деятельность ортопедов направлена на устранение деформаций верхних и нижних конечностей, восстановление их функциональности и качественной подвижности.
Продолжительность жизни пациентов, рождённых с диагнозом «акрофациальный дизостоз» (синдром Нагера) составляет от 20 до 50 лет. Зависит она от состояния общего здоровья, степени функциональности внутренних органов и тяжести заболевания.
Учитывая отсутствие нарушений в психомоторном развитии, можно смело сказать, что пациенты с синдромом Нагера способны вести активную жизнь, несмотря на необходимость создания специальных условий для некоторых из них.
Чаще всего жизненные трудности связаны с нарушением слуха, но регулярные занятия с сурдопедагогом помогают пациентам наладить контакт с окружающими. Своевременно проведённая операция по вживлению слухового аппарата позволит малышу избежать отставания в речевом и психическом развитии.
Сложность представляют случаи, когда врачам приходится сталкиваться с самой тяжёлой 4 степенью нарушения формирования и развития лучевой кости, гортани и надгортанника, а также с:
- отсутствием оболочки органов зрения;
- полным сращением пальцев стопы;
- укорочением конечностей.
Потребуется масса усилий и длительное лечение, чтобы помочь пациенту стать полноценным членом общества. Современные пластические и челюстно-лицевые хирурги, ортопеды, стоматологи и другие специалисты способны устранить большую часть дефектов в развитии малыша.
Проводя сложные операции, медики добиваются восстановления функциональности не только конечностей, но и внутренних органов. Хирургическое вмешательство проводится детишкам с синдромом Нагера сразу после появления на свет. Это даёт им возможность свободно дышать и глотать. Отсутствие отставания в других сферах развития обеспечивает таким пациентам нормальную продолжительность жизни.
Чтобы уберечь будущего ребёнка от столь серьёзного заболевания, нужно заранее пройти обследование, получить консультацию опытного генетика, вести здоровый образ жизни, заботясь о собственном здоровье и о здоровье долгожданного малыша.
Источник
Дифференциальный диагноз ахондроплазии у плода
При дифференциальной диагностике следует учитывать многие состояния:
• танатофорная дисплазия (узкая грудная клетка, уплощение позвонков, более выраженная микромелия, отсутствие признака «рука-трезубец», более выраженное многоводие);
• ахондрогенез (выраженное нарушение минерализации с отсутствием гиперэхогенности костей, более выраженные микромелия и многоводие);
• несовершенный остеогенез II типа (нарушение минерализации костей: во многих случаях имеется возможность хорошо визуализировать проксимальные по отношению к датчику отделы мозга через «прозрачные» кости свода черепа, различные степени микромелии, обнаружение в некоторых случаях угловых деформаций костей вследствие переломов, колоколообразная грудная клетка);
• диастрофическая дисплазия (при которой микромелия сочетается с контрактурами суставов с наибольшей выраженностью аномальности положения пальцев кистей и стоп).
У детей с ахондроплазией отмечается нормальное умственное развитие. Основные проблемы являются ортопедическими (сужение спинномозгового канала и большого затылочного отверстия). Существует активно действующая группа поддержки таких пациентов (Маленькие люди Америки — The Little People of America), которая принимает очень большое участие в разрешении проблем таких больных.
Акушерская тактика. Несмотря на то что пациентке может быть предложено прерывание беременности до наступления периода жизнеспособности плода, большинство таких детей хорошо адаптируются в обществе и ведут продуктивную жизнь.
Ахондроплазия у плода
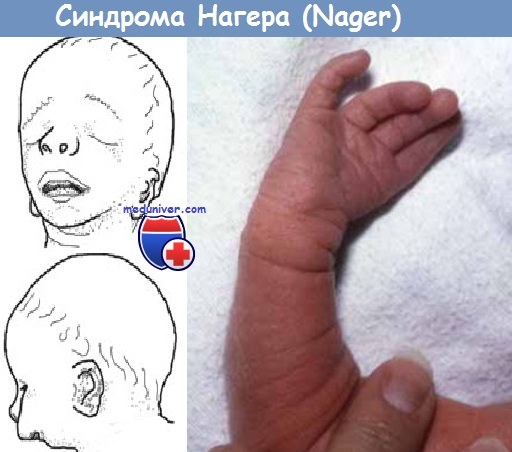
Синдром акрофациального дизостоза — синдром Нагера
Определение. Синдром Нагера (Nager) включает в себя аномалии конечностей в виде аплазии лучевой кости, синостоза лучевой и локтевой костей, аплазии или гипоплази большого пальца кисти, выраженной микрогнатии и гипоплазии маляров.
Синонимы. Мандибулофациальный дизостоз типа Тричера-Коллинза (Treacher-Collins) с аномалиями конечностей; акрофациальный дизостоз Нагера (Nager). Вариантом этого заболевания является синдром летального акрофациального дизостоза Родригеса (Rodriguez), который сопровождается недоразвитием преаксиальных и аномалиями постаксиальных отделов конечностей, а также выраженной гипоплазией плечевого и тазового пояса, пороками сердца и центральной нервной системы (ЦНС).
Распространенность. Встречаются не часто.
Этиология. Синдром Нагера (Nager) может возникать в результате новых аутосомно-доминантных мутаций.
Диагностика. Диагностика основывается на выявлении выраженной микрогнатии и типичной микромезомелии с аномалиями развития верхних конечностей, на основании которых возможно установление диагноза. Описано частое наличие других сочетанных аномалий.
Генетические нарушения. Гены, ответственные за возникновение синдрома Нагера (Nager), локализуются на длинном плече хромосомы 9q32.
Дифференциальный диагноз. Другие заболевания, при которых имеется микрогнатия и дистальная эктромелия, в частности, трисомия 18.
Прогноз. Заболевание является летальным вследствие гипоплазии легких, возникающей из-за выраженной гипоплазии нижней челюсти.
Акушерская тактика. До наступления периода жизнеспособности плода может быть предложено прерывание беременности. Если принимается решение о ее пролонгировании, стандартная тактика пренатального ведения пациентки не изменяется. Большое значение для генетического консультирования семейной пары имеет подтверждение диагноза после рождения.
— Также рекомендуем «Акромезомелическая дисплазия. Синдром Эйкарди»
Оглавление темы «Врожденные пороки развития плода»:
1. Особенности диагностики фетальных синдромов. Ахондрогенез и его частота
2. Генетические нарушения при ахондрогенезе. Ахондроплазии у плода
3. Дифференциальный диагноз ахондроплазии у плода. Синдром акрофациального дизостоза — синдром Нагера
4. Акромезомелическая дисплазия. Синдром Эйкарди
5. Врожденный СПИД. Фетальная ВИЧ-инфекция
6. Синдром амниотических перетяжек. Диагностика и прогноз при синдроме амниотических перетяжек
7. Синдром Апера у плода. Диагностика и прогноз при синдроме Апера
8. Синдром Арнольда-Киари. Множественный врожденный артрогрипоз плода
9. Асфиксическая дисплазия грудной клетки. Синдром асплении и полисплении
10. Синдром Беквита-Видемана. Диагностика и прогноз синдрома Беквита-Видемана
Источник
Мико, где то я уже видела это видео… и по моему этот «придурок» ее отец, который ее растит и пытается по возможности развивать. А ты смог бы, если б у тебя такая дочь родилась? Тот же вопрос кстати и к Василию…
Я бы это чучело в мусопровод выкинул, вместе с мамашкой дефектной, которая не может нормального ребенка родить. Что это за хрень? Она же до конца свой, надеюсь непродолжительной жизни, будет мучиться и других мучить
Дина, только выглядит он здесь точно, как «придурок», на фоне его ангельской, милой внешности, несчастная девочка выглядит еще уродливей….. на вопрос отвечу, никогда бы не смогла выставлять такую дочь, тем более в грязном интернете……. не бросила бы, но и никогда и никому не показала…..
Серафим, а откуда такая уверенность, что дело именно в «мамашке»? Может, дефектные гены у ребенка от отца? В создании ребенка оба родителя участвую на равных, в нем от каждого поровну. Вот с ребенком и целесообразностью такой жизни уже вопрос, и это зависит от многих факторов.
Серафим, кхм, вместе с «мамашкой»?? Причём здесь она? Есть много факторов определяющих все эти деффекты!
Дефекты, с одной «ф»)
какие такие факторы? Если уродца родила, значит больная, у здоровых людей здоровые дети рождаются, нормальные, а не вот такое непойми что
Серафим, зря тебя, вместе с твоей мамашей не выкинули…. воспитать такого выродка, как ты, еще постараться нужно….
![]()
Серафим, тебе бы генетику повторить бы
Хлебало завали, иди лучше посуду помой
А в ком же еще? У отца дело маленькое, сунул, плюнул и пошел. Если урод рождается мать виновата
Серафим, нырни обратно в ту дыру, откуда ты всплыл
Дина, это не её отец, а человек с канала Special Books by Special Kids 🙂
Серафим, да неужели? «Предположительно заболевание носит аутосомно-доминантное наследование. Что это значит? Мутантный ген, который передаёт заболевание, есть у обоих родителей. Вероятность рождения больного ребёнка составляет ровно 50%, то есть он будет либо полностью здоров, либо болен. Носительства мутантного гена без появления признаков болезни здесь нет. Заболевание может проявиться как у мальчиков, так и у девочек.»
Учите генетику.
Я не верю в эти сказки и вам не советую
Серафим, я тоже хотела вам это посоветовать)))
Серафим, да, действительно!
У меня вопрос что у тебя было с биологией??
Первый главный фактор это окружающая среда и проблемы экологии.
И даже если женьщина была здоровая как бык и беременность протикала отлично, но тут она случайно простужается по вине своего мужа, например. И если она не сможет вылечиться до осложений, может получиться именно этот дефект у ребёнка, а так же «заячья губа».
![]()
Серафим, при здоровых родителях могут рождаться такие дети, если кто-то из них является носителем этого гена, как с мужской стороны, так и с женской
Показать следующие комментарии
Источник
Синдром Нагера – Причины Симптомы и Лечение
Человечеству известно немало врожденных аномалий, однако и сегодня на свет появляются детишки с жуткими генетическими отклонениями, причины которых неизвестны, а способы лечения сложны и не всегда эффективны. К таким аномалиям относится и акрофациальный дизостоз, более известный как синдром Нагера. Подробнее об этом тяжелом генетическом отклонении расскажем в данной статье.
Что необходимо знать о заболевании
Синдром Нагера или акрофациальный дизостоз – редкое наследственное заболевание, которое впервые было описано в 1948 году двумя швейцарскими учеными – Ж. де Рейнером и Феликсом Нагером. При нем на свет появляются дети с ужасными черепно-лицевыми и опорно-двигательными аномалиями. На сегодняшний день во всем мире зафиксировано не более 300 случаев рождения детей с рассматриваемым синдромом.
Причины аномалии
Болезнь предположительно имеет аутосомно-доминантное наследование, то есть появляется лишь в том случае, когда развивающийся в утробе плод получает мутировавший ген от обоих родителей. В этом случае вероятность появления на свет ребенка с отклонениями в развитии равна 50%. Причем аномалии в равной степени подвержены и мальчики, и девочки.
Тем не менее, медицине известны случаи так называемой спонтанной мутации, когда детишки с синдромом Нагера появлялись у вполне здоровых родителей. Что стало причиной мутации в этом случае, остается загадкой даже для светил науки. Тем не менее, к факторам, которые могут спровоцировать развитие акрофациального дизостоза, специалисты относят:
- проживание в районах загрязненных радиацией или вблизи мест добычи радиоактивных металлов;
- вредные привычки родителей (наркомания, токсикомания, алкоголизм);
- облучение матери во время беременности.
Ученым удалось установить, что мутировавший ген расположен в хромосоме с названием 9q32.
Симптомы синдрома Нагера
Диагноз плоду, имеющему такое тяжелое заболевание, устанавливается просто, причем еще на ранней стадии беременности. Для этого врачу достаточно заметить минимальные проявления хромосомной аномалии, а именно:
- недоразвитие нижней челюсти или полное отсутствие челюстных суставов;
- стеноз (сужение) наружного слухового прохода или полное отсутствие такового. Боле того, для синдрома Нагера характерно нарушение строения всего слухового аппарата, а значит дети, появляющиеся на свет с таким диагнозом, чаще всего абсолютно глухие;
- антимонголоидный разрез глаз, при котором заметно опущение наружных краев глаза и наблюдается опущение верхних век;
- недоразвитие или полное отсутствие лучевых костей (костей проходящих от запястья до локтя). Детишки, рождающиеся с акрофациальным дизостозом, как правило, имеют изогнутые лучевые кости, из-за которых кисти рук находятся в неестественном положении;
- недоразвитие или отсутствие пальцев на кистях рук.
В подавляющем большинстве случаев медики сталкиваются с 4 степенью недоразвития лучевой кости и отсутствием первых двух лучей кисти (костей мизинца и безымянного пальца). Тем не менее, часто встречаются пациенты, у которых все кости пальцев на руках присутствуют, однако наблюдается серьезное искривление мизинца и ограничение движения руки в локте.
Кроме того, у различных пациентов с синдромом Нагера отмечаются и другие аномальные отклонения, а именно: дефицит роста, гипоплазия гортани и надгортанника, отсутствие оболочки глаза, отсутствие или недоразвитость пальцев ступни, вальгусный большой палец, косолапость и укорочение конечностей. Из других проявлений заболевания у больных встречаются: аномалии шейного отдела позвоночника, сколиоз, гидроцефалия, микроцефалия, некоторые мочеполовые аномалии, стеноз ликворных путей, пороки полушарий головного мозга, болезнь Гиршпрунга и уртикарная сыпь. Стоит заметить, что перечисленные проявления синдрома Нагера наблюдаются не у всех больных.
В некоторых случаях младенцы с синдромом Нагера имеют врожденные пороки сердца (чаще всего тетрада Фалло). Некоторые аномалии сердца и сосудов несовместимы с жизнью.
Что касается умственной отсталости таких пациентов, то в большинстве случаев и мальчики, и девочки с акрофациальным дизостозом имеют нормальный интеллект. При этом двусторонняя глухота и челюстные аномалии у таких пациентов провоцируют проблемы с артикуляцией и замедляют психомоторное развитие больных.
Дифференциальный диагноз
Стоит заметить, что синдром Нагера имеет много общего с такими тяжелыми патологиями развития, как:
- синдром Гольденхара;
- синдром Тричера-Коллинза;
- синдром Миллера;
- синдром Халлермана-Штрайфа.
Чтобы поставить правильный диагноз, специалисту необходимо провести внешний осмотр и УЗИ-исследование. Кроме того, в обязательном порядке проводятся консультации с ЛОР-врачом, урологом, стоматологом, окулистом и другими узкими специалистами.
Прерывание беременности
Наличие синдрома Нагера и тяжесть заболевания диагностируется при помощи ультразвукового исследования уже после 16 недель беременности. В этом случае будущей матери необходимо пройти генетическую консультацию и решить вопрос – вынашивать ребенка или согласиться на медицинское прерывание беременности, т.е. на аборт. Если родители решают вынашивать ребенка, далее беременность ведет акушер-гинеколог без каких-либо особенностей.
Возможно ли лечение синдрома Нагера
Сразу скажем, что акрофациальный дизостоз является врожденной генетической аномалией, а значит, неизлечимой болезнью. Тем не менее, ребенку можно помочь, ведь хирургические операции, при которых врачи исправляют дефекты челюсти, ушей, а также пальцев рук и ног, на сегодняшний день не редкость. Кроме того, таким пациентам могут проводиться операции по устранению дефектов внутренних органов.
Однако одних пластических операций детишкам с синдромом Нагера недостаточно. Как правило, у них наблюдается двусторонняя глухота, а значит, кроме исправления челюсти и рук, больным требуется установка слухового протеза. Специалисты рекомендуют проводить слухопротезирование в раннем возрасте, чтобы ребенок мог избежать проблем с интеллектуальным развитием.
Дефект речи – еще одна проблема, с которой сталкиваются пациенты с акрофациальным дизостозом. Для решения проблемы им требуются занятия с сурдопедагогом.
Прогноз
На сегодняшний день прогноз на жизнь пациентов с рассматриваемым диагнозом в основном благоприятный. Раньше из появившихся на свет детишек с синдромом Нагера выживало не более 25%, а все потому, что из-за гипоплазии нижней челюсти новорожденный не мог дышать и глотать. Сегодня же таких детишек оперируют непосредственно при рождении, исправляя этот недостаток.
В остальном же дети с синдромом Нагера не имеют отставания в развитии, а продолжительность жизни у них такая же, как и у здоровых детей. Для нормальной жизни и полноценного развития таким пациентам требуется помощь узких специалистов (сурдопедагога) и психологическая поддержка родителей.
Берегите своих детей!
Источник