Синдром нагера что это фото

Главная » Болезни и мутации
|
27.06.2019
: 18072
: 0

????
операция, хирургия, лицо, аномалия, Синдром Нагера
6-летняя Дарина Шпенглер из Красноярского края родилась с аномалией, из-за которой у нее отсутствуют губы и подбородок. В целом вся нижняя часть лица у нее не развита. Диагноз девочке официально никогда не был поставлен, но предполагают, что у нее редкий врожденный Синдром Нагера.
Мать ничего не знала об этой аномалии до родов, а когда Дарина родилась, медсестры долго не показывали ребенка матери — Елене Шпенглер. Когда же они наконец принесли ей девочку, то мать упала в обморок, едва взглянув на нее.
Отец же Дарины был совершенно невозмутим, когда увидел ребенка. «Его лицо нисколько не изменилось», — рассказывает Елена.

Когда врачи стали рекомендовать Елене отказаться от ребенка, мать не стала их слушать. Впоследствии из-за состояния Дарины родители девочки разругались с большинством своих родственников (те пеняли им, что не оставили ребенка в больнице) и уехали из деревни, где жили.
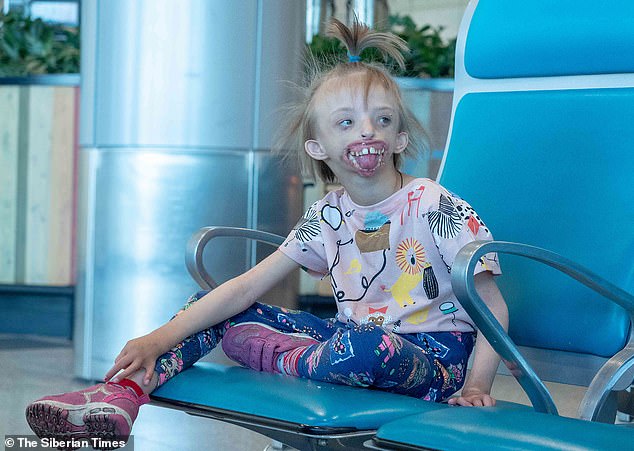
Когда Дарина была младше она много плакала, потому что ткани вокруг челюстей постоянно кровоточили и болели.

Несколько лет родители девочки пытались найти помощь у российских хирургов, однако им никто не смог помочь. Надежда проснулась, когда появился шанс поехать в Великобританию в знаменитую лондонскую детскую больницу на Грейт Ормонд -стрит.
Из-за открытых челюстей Дарине очень сложно есть, поэтому в свои шесть лет она весит ниже нормы и на вид ей не дашь больше четырех. После операции, которая закроет челюсти, она сможет наконец питаться как все люди.
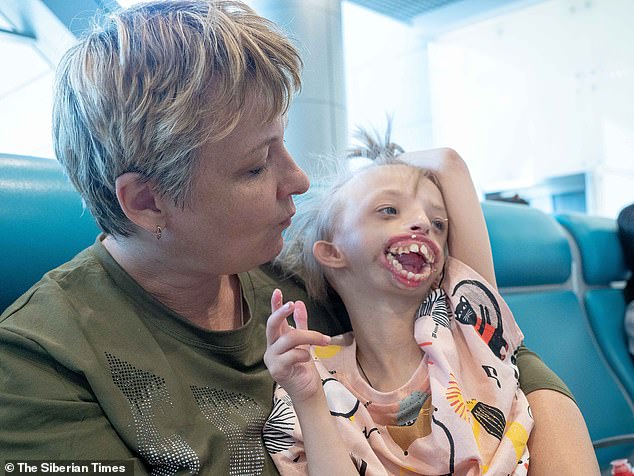
Но больше всего родители девочки надеются на то, что во время операции ей исправят и голосовые связки, ведь Дарина пока не может произнести ни слова.
Внешность Дарины шокирует многих неподготовленных людей. Прохожие на улицах смотрят ей вслед, а некоторые даже советуют родителям девочки надевать на Дарину маску. Из-за этого друзей среди детей у Дарины нету, дети боятся играть с ней.

Однажды родителей девочки даже обвинили в том, что они сами нанесли ей эти раны. Им звонила полиция и требовала рассказать, что произошло. Лишь после всех объяснений полиция перестала их беспокоить.

Дарина имеет нормальное умственное развитие и уже понимает, что с ней что-то не так. Родители переживают из-за этого, но отказываются прятать девочку от общества и тем более надевать на нее маску. Они принимают ее такой, какая она есть, даже несмотря на то, что девочку из-за ее внешности не приняли ни в один детский сад. Пару раз в неделю на дом к Дарине приходят учителя и учат ее читать и писать.
Дарина и ее мама Елена уже прилетели в Лондон и сейчас готовятся к операции. Стоимость операции равна 85 тысячам долларов и оплачивается российскими благотворителями через общество Русфонд.

Синдром Нагера влияет на лицевые кости и он настолько редок, что в мире зафиксировано лишь около 100 таких случаев.
![]() Если с вами произошел необычный случай, вы увидели странное существо или непонятное явление, вам приснился необычный сон, вы увидели в небе НЛО или стали жертвой похищения пришельцев, вы можете прислать нам свою историю и она будет опубликована на нашем сайте
Если с вами произошел необычный случай, вы увидели странное существо или непонятное явление, вам приснился необычный сон, вы увидели в небе НЛО или стали жертвой похищения пришельцев, вы можете прислать нам свою историю и она будет опубликована на нашем сайте
===>
Прислать историю
.
Новое в Блогах и Форуме
Источник
Эта статья создана пользователем Онедио. Со стороны редакции изменений не было. Вы тоже можете создавать свои статьи на нашем сайте.
Onedio избранное > Интересно-16 января 2018, 09:57‘ добавлено,
17 января 2018, 11:23‘ обновлено
Наверняка у вас тоже есть недостатки, которых вы стесняетесь и которые всячески стараетесь исправить… Не переживайте слишком сильно по этому поводу, ведь у некоторых людей такие недостатки имеют гораздо более серьёзное происхождение (да и их и недостатками уже назвать нельзя, это самые что ни на есть настоящие уродства). Посмотрим?
Источник: https://list25.com/25-people-with-strang…
1. Семья Улас
list25.com
Эта семья из Турции всё время передвигается на четырёх конечностях, а всё из-за редкого вида инвалидности. Людям просто не хватает равновесия, чтобы держаться на двух ногах.
2. Хосе Местре
Показать изображение
list25.com
Опухоль весом в 5.5 кг буквально целиком съела лицо португальца Хосе Местре. Такое уродство вызвало сильный рост кровеносных сосудов, который ослепил мужчину, добрался до его рта (затрудняя речь и мешая есть). Хирург из Чикаго взялся за сложнейшую операцию по удалению опухоли, и ему это успешно удалось.
3. Неизвестный
list25.com
Звучит иронично, но у людей действительно могут расти рога. Всё из-за излишнего количества кератина, который вырабатывается кожей. К счастью, такие рога можно удалить хирургическим путём…
4. Бри Уолкер
Телеведущая из Лос-Анджелеса живёт с врождённым пороком под названием эктродактилия («клешнеобразная кисть»). Порок заключается в недоразвитии одного или нескольких пальцах на кистях или стопах.
5. Хавьер Ботет
Этого мексиканского актёра также называют ходячим спецэффектом, ведь именно его зовут сниматься во многих ужастиках. А всё из-за врождённого генетического заболевания, которое называется «болезнь Марфана» или «Синдром Марфана». Люди с таким заболеванием отличаются неестественной худобой, высоким ростом, а также удлинёнными конечностями и пальцами.
6. Петеро Бьякатонда
list25.com
Петеро родом из небольшого городка в Уганде и страдает от врождённой аномалии развития черепа под названием синдром Аперта. У страдающих этим заболеванием наблюдается отсталость умственного развития, выпуклый лоб, вогнутое лицо, а также деформация челюсти.
7. Руди Сантос
Показать изображение
list25.com
Когда Руди был в утробе матери, у него был брат-близнец, но затем Руди просто его поглотил, в результате чего начал страдать от паразитарной краниопаги (особый тип сращения сиамских близнецов). Сейчас у Руди дополнительная пара ног и рук
8. Харри Реймонд Истлек
list25.com
Харри страдал от очень редкого недуга под названием прогрессирующая оссифицирующая фибродисплазия (ФОП). При этом у человека соединительные ткани начинают превращаться в кости, в результате чего формируется буквально новая структура скелета.
9. Деде Косвара
Показать изображение
list25.com
Этого индонезийца также называют «человеком-деревом», ведь всё его тело покрывают ужасные наросты в виде бородавок. Иммунная система этого мужчины не может побороть такой активный рост бородавок. К сожалению, в 2016 году мужчина скончался.
10. Дидье Монтальво
list25.com
Этот мальчик из Колумбии страдает заболеванием, из-за которого по всему его телу растут родинки. Одна из них (на спине) стала настолько большой, что напоминает панцирь черепахи. Слава Богу, что британскому хирургу удалось помочь мальчику, удалив родинку, и теперь он живёт нормальной жизнью.
11. Тесса Эванс
list25.com
Девочка страдает от аплазии — врождённый порок, который заключается в отсутствии какой-либо части тела (в данном случае носа). К счастью, таким детям можно прибегать к протезированию, но вот запахов Тесса слышать уже никогда не будет.
12. Дин Эндрюс
list25.com
Дин страдает от редкого генетического дефекта под названием «прогерия». Он заключается в преждевременном старении организма. Сейчас Дину всего 20, но выглядит он на 100 лет!
13. Неизвестная
list25.com
Синдром Тричера Коллинза — это аутосомно-доминантное заболевание, характеризующееся черепно-лицевой деформацией. Оно может повлиять на развитие скул, челюсти, подбородка и ушей человека. В некоторых случаях дефекты можно исправить пластическими операциями.
14. Деклан Хайтон
list25.com
Этот мальчик из Великобритании страдает от врождённого Синдрома Мёбиуса, из-за которого он не может двигать мышцами лица (в том числе глазами).
15. Верн Тройер
list25.com
Этот американский актёр страдает от дисплазии — генетической болезни, которая передалась ему от отца. Поэтому его рост равен всего 80 см.
16. Манар Магед
list25.com
Как и Руди Сантос, Манар Магед страдает от от паразитарной краниопаги. Но хирурги не смогли разделить близнецов, поэтому девочка скончалась.
Эта статья создана пользователем Онедио. Со стороны редакции изменений не было. Вы тоже можете создавать свои статьи на нашем сайте.
Источник
Мико, где то я уже видела это видео… и по моему этот «придурок» ее отец, который ее растит и пытается по возможности развивать. А ты смог бы, если б у тебя такая дочь родилась? Тот же вопрос кстати и к Василию…
Я бы это чучело в мусопровод выкинул, вместе с мамашкой дефектной, которая не может нормального ребенка родить. Что это за хрень? Она же до конца свой, надеюсь непродолжительной жизни, будет мучиться и других мучить
Дина, только выглядит он здесь точно, как «придурок», на фоне его ангельской, милой внешности, несчастная девочка выглядит еще уродливей….. на вопрос отвечу, никогда бы не смогла выставлять такую дочь, тем более в грязном интернете……. не бросила бы, но и никогда и никому не показала…..
Серафим, а откуда такая уверенность, что дело именно в «мамашке»? Может, дефектные гены у ребенка от отца? В создании ребенка оба родителя участвую на равных, в нем от каждого поровну. Вот с ребенком и целесообразностью такой жизни уже вопрос, и это зависит от многих факторов.
Серафим, кхм, вместе с «мамашкой»?? Причём здесь она? Есть много факторов определяющих все эти деффекты!
Дефекты, с одной «ф»)
какие такие факторы? Если уродца родила, значит больная, у здоровых людей здоровые дети рождаются, нормальные, а не вот такое непойми что
Серафим, зря тебя, вместе с твоей мамашей не выкинули…. воспитать такого выродка, как ты, еще постараться нужно….
![]()
Серафим, тебе бы генетику повторить бы
Хлебало завали, иди лучше посуду помой
А в ком же еще? У отца дело маленькое, сунул, плюнул и пошел. Если урод рождается мать виновата
Серафим, нырни обратно в ту дыру, откуда ты всплыл
Дина, это не её отец, а человек с канала Special Books by Special Kids 🙂
Серафим, да неужели? «Предположительно заболевание носит аутосомно-доминантное наследование. Что это значит? Мутантный ген, который передаёт заболевание, есть у обоих родителей. Вероятность рождения больного ребёнка составляет ровно 50%, то есть он будет либо полностью здоров, либо болен. Носительства мутантного гена без появления признаков болезни здесь нет. Заболевание может проявиться как у мальчиков, так и у девочек.»
Учите генетику.
Я не верю в эти сказки и вам не советую
Серафим, я тоже хотела вам это посоветовать)))
Серафим, да, действительно!
У меня вопрос что у тебя было с биологией??
Первый главный фактор это окружающая среда и проблемы экологии.
И даже если женьщина была здоровая как бык и беременность протикала отлично, но тут она случайно простужается по вине своего мужа, например. И если она не сможет вылечиться до осложений, может получиться именно этот дефект у ребёнка, а так же «заячья губа».
![]()
Серафим, при здоровых родителях могут рождаться такие дети, если кто-то из них является носителем этого гена, как с мужской стороны, так и с женской
Показать следующие комментарии
Источник
Синдром Тричера Коллинза (англ. Treacher Collins syndrome, TCS, челюстно-лицевой дизостоз) — аутосомно-доминантное заболевание, характеризующееся черепно-лицевой деформацией. Описан английским офтальмологом Эдвардом Тричером Коллинзом в 1900 году[1].
Синдром Тричера Коллинза встречается у 1 из 50 000 младенцев[2]. Типичные клинические признаки: грубый дефект лицевой части черепа, косоглазие, колобомы век; размер рта, подбородка и ушей существенно меньше нормы. В некоторых случаях — ослабление слуха.
Этиология[править | править код]
Причиной заболевания является, чаще всего, нонсенс-мутация (возникновение стоп-кодона) в гене TCOF1, приводящая к гаплонедостаточности.
Ген TCOF1 расположен в локусе 5q32-33, на длинном (q) плече 5-й хромосомы, начинается от 149 737 202-й пары оснований и заканчивается на 149 779 871-й паре оснований[3][4].
Продукт гена TCOF1 — ядерный транспортный белок, который экспрессируется во многих тканях во время эмбрионального и постэмбрионального развития и принимает участие в транскрипции ДНК. При синдроме Тричера Коллинза развивается состояние, при котором половинного количества генного продукта недостаточно для нормального функционирования организма[5].
Синдром наследуется по аутосомно-доминантному принципу и характеризуется высокой пенетрантностью. Экспрессивность может быть от умеренной до выраженной, потому тяжесть дефекта отлична у разных пациентов — от почти незаметных признаков до крайне тяжёлых форм. У большинства пациентов слаборазвитые лицевые кости, что приводит к «затонувшему» лицу, крупный нос и очень маленькие челюсти и подбородок (микрогнатия). У некоторых больных присутствует волчья пасть. В тяжелых случаях микрогнатия может вытеснять язык пострадавших новорожденных достаточно, чтобы вызвать преграду ротоглотки и потенциально опасных для жизни заболеваний дыхательных путей. Врождённый порок сердца является необычной особенностью[6].
Лечение[править | править код]
В лечении пациентов, пострадавших от ТКС, применяется междисциплинарный подход, то есть требуются вмешательства различных специалистов. Основные проблемы у пациентов с ТКС — нарушения глотания и проходимости дыхательных путей. Некоторые пациенты нуждаются в трахеостомии. Гастростома может быть необходима для обеспечения адекватного потребления пищи и для защиты дыхательных путей[7]. Вопрос о хирургическом восстановлении структуры лица решается индивидуально и осуществляется по достижении определённого возраста[8].
Потеря слуха при синдроме Тричера Коллинза вызвана деформацией структур в наружном и среднем ухе. Потеря слуха, как правило, двусторонняя. Даже в тех случаях, когда ушные раковины и наружные слуховые проходы не затронуты синдромом, цепи слуховых косточек часто повреждены[9].
Попытки хирургическим путём восстановить наружный слуховой проход для улучшения слуха у детей с ТКС не дали положительных результатов[10]. Предпочтительной оказалась слуховая реабилитация со слуховыми аппаратами костной проводимости[en] (BAHA) или обычными слуховыми аппаратами[11].
В культуре[править | править код]
- «Чудо» — художественный фильм-драма 2017-го года с главным героем с синдромом ТК
Примечания[править | править код]
- ↑ Treacher Collin E, «Cases with symmetrical congenital notches in the outer part of each lid and defective development of the malar bones», 1900, Trans Ophthalmol Soc UK, 20, p. 190—192
- ↑ Chiara Conte, Maria Rosaria D’Apice, Fabrizio Rinaldi, Stefano Gambardella, Federica Sangiuolo and Giuseppe Novelli. Novel mutations of TCOF1 gene in European patients with Treacher Collins syndrome (англ.) // BMC Med Genet. : journal. — 2011. — 27 September (vol. 12). — doi:10.1186/1471-2350-12-125. — PMID 21951868.
- ↑ Dixon M.J., Dixon J., Raskova D., et al. Genetic and physical mapping of the Treacher Collins syndrome locus: refinement of the localization to chromosome 5q32-33.2 (англ.) // Human Molecular Genetics (англ.)русск. : journal. — Oxford University Press, 1993. — Vol. 1, no. 4. — P. 249—253. — doi:10.1093/hmg/1.4.249. — PMID 1303194.
- ↑ Dixon M.J., Dixon J., Houseal T., et al. Narrowing the position of the Treacher Collins syndrome locus to a small interval between three new microsatellite markers at 5q32-33.1 (англ.) // American Journal of Human Genetics (англ.)русск. : journal. — 1993. — Vol. 52, no. 5. — P. 907—914. — PMID 8488840.
- ↑ Синдром Тричер Коллинза-Франческетти (мандибулофациальный дизостоз, TCOF) //Центр молекулярной генетики Медико-генетического научного центра РАМН
- ↑ Электронный научный журнал «Медицина и образование в Сибири»
- ↑ Goel L., Bennur S.K., Jambhale S. Treacher Collins syndrome-a challenge for anaesthesiologists (англ.) // Indian Journal of Anaesthesia (англ.)русск. : journal. — 2009. — August (vol. 53, no. 4). — P. 496—500. — PMID 20640217.
- ↑ Evans, Adele Karen; Rahbar, Reza; Rogers, Gary F.; Mulliken, John B.; Volk, Mark S. Robin sequence: A retrospective review of 115 patients (англ.) // International Journal of Pediatric Otorhinolaryngology : journal. — 2006. — 31 May (vol. 70, no. 6). — P. 973—980. — doi:10.1016/j.ijporl.2005.10.016. — PMID 16443284.
- ↑ Argenta, Louis C.; Iacobucci, John J. Treacher Collins Syndrome: Present concepts of the disorder and their surgical correction (англ.) // World Journal of Surgery (англ.)русск. : journal. — 1989. — 30 June (vol. 13, no. 4). — P. 401—409. — doi:10.1007/BF01660753. — PMID 2773500.
- ↑ Marres, HA; Cremers, CW; Marres, E.H. Treacher-Collins syndrome. Management of major and minor anomalies of the ear (англ.) // Revue de laryngologie — otologie — rhinologie : journal. — 1995. — Vol. 116, no. 2. — P. 105—108. — PMID 7569369.
- ↑ Marres, H.A. Hearing loss in the Treacher-Collins syndrome (неопр.) // Advances in oto-rhino-laryngology. — 2002. — Т. 61. — С. 209—215. — doi:10.1159/000066811. — PMID 12408086.
Ссылки[править | править код]
- «Новое лицо Джулианы» — документальный фильм о девочке, родившейся с синдромом Тричера Коллинза тяжёлой формы
Источник

Синдром Аксенфельда-Ригера – патология, при которой поражение органа зрения сочетается с системными мальформациями. Глазные проявления включают тяжелую степень дисгенеза угла передней камеры. К числу системных аномалий относятся пороки развития зубов, костей черепа и внутренних органов. Диагностика направлена на проведение гониоскопии, биомикроскопии, тонометрии глаза, компьютерной периметрии, офтальмоскопии. Лечение сводится к инстилляциям гипотензивных средств из группы ингибиторов КАГ, альфа-агонистов, бета-блокаторов, а при низкой эффективности – проведению трабекулоэктомии.
Общие сведения
Синдром Аксенфельда-Ригера — это редкое заболевание, развитие которого генетически детерминировано. Однако, только у 40% больных удается обнаружить хромосомную аномалию и идентифицировать поврежденный ген. Болезнь названа в честь немецкого офтальмолога Т. Аксенфельда и австрийского ученого Г. Ригера. Показатель заболеваемости находится в пределах 1:200 000. Повышение внутриглазного давления, которое манифестирует в клинику вторичной глаукомы встречается более, чем в 50% случаев. Патология с одинаковой частотой встречается среди лиц мужского и женского пола.

Причины синдрома Аксенфельда-Ригера
Этиология заболевания до конца не изучена. Основная теория развития синдрома Аксенфельда-Ригера – генетическая. Выявлены мутаций двух генов, ассоциированных с возникновением болезни. Тип наследования — аутосомно-доминантный, но наблюдаются спорадические случаи. Это является основанием полагать, что существует и другие причины, которые на сегодняшний день неизвестны.
Патогенез
Доказано, что ген FOXC1 (хромосомы 6p25) и PITX2 (хромосомы 4q25) кодируют белки, которые действуют как факторы транскрипции и регулируют экспрессию генов. При мутации гена PITX2 нарушается гистогенез глаз, зубов и брюшной полости. FOXC1 отвечает за формирование переднего сегмента глазного яблока. Ученые полагают, что изменения в локусе хромосомы 13q14 также играют важную роль в патогенезе, однако эта теория еще не подтверждена.
При гистологическом исследовании было выявлено, что для синдрома Аксенфельда-Ригера характерно нарушение миграции клеток нервного гребня, которые в норме формируются по периферии эмбриональной нервной пластинки еще до момента закрытия нервной трубки. Это лежит в основе дисгенеза УПК, что может стать причиной повышения внутриглазного давления. Гипоплазия экстраокулярных мышц обусловлена нарушением дифференциации мезодермального комплекса.
Симптомы синдрома Аксенфельда-Ригера
Для заболевания характерно двухстороннее течение, однако изменения чаще асимметричны. Родители еще в период новорожденности обращают внимание на косметический дефект, который обусловлен аномальным строением радужной оболочки. Длительное время для синдрома Аксенфельда-Ригера свойственно стабильное течение. Поводом для беспокойства является ухудшение зрения, которое возникает следом за повышением ВГД.
К числу краниофациальных признаков относят гипоплазию верхней челюсти, уменьшение зубов в размере (микродонтия) или отсутствие отдельных зубов (чаще верхнечелюстных молочных или постоянных центральных резцов). В периумбиликальной зоне выявляется дополнительная складка кожи. Нередко отмечаются аномалии строения паховой области, ухудшение слуха, задержка умственного развития. У мальчиков может возникать гипоспадия.
Осложнения
Синдром Аксенфельда-Ригера в большинстве случаев сопровождается офтальмогипертензией. Признаки вторичной глаукомы зачастую выявляются еще в юношеском возрасте. Пациенты подвержены риску отслойки сетчатки в связи с пролиферативной витреоретинопатией. Патология часто сочетается с косоглазием, которое сложно поддается коррекции. Наиболее неблагоприятное осложнение синдрома – фтизис глазного яблока.
Диагностика
Для подтверждения диагноза рекомендовано собрать анамнез, провести физикальное обследование, осмотр лица и ротовой полости, генетическое исследование (прямое секвенирование кодирующей области генов FOXC1 и PITX2). Выполнение пренатального тестирования целесообразно при наличии в семье или у кровных родственников синдрома Аксенфельда-Ригера. Основные методы диагностики:
- Гониоскопия. Визуализируется задний эмбриотоксон. Вдоль проминирующей линии Швальбе видны отложения пигмента. Характерна адгезия к структурам УПК отростков, идущих от периферии радужки. В отдельных сегментах они достигают линии Швальбе и трабекулярной сети.
- Биомикроскопия глаза. Определяются признаки гипоплазии стромы радужки, которые наиболее выражены в области цилиарного пояса. Выявляется истончение поверхностного листка радужной оболочки и отек роговицы. Болезнь сопровождается легкой сегментарной гипоплазией. Реже наблюдается корэктопия и псевдополикория.
- Измерение внутриглазного давления. Измерение ВГД позволяет в 50% пациентов выявить признаки офтальмогипертензии. При этой патологии очень важно оценивать результаты тонометрии с учетом толщины роговицы. Данные пахиметрии в отдельных случаях указывают на снижение ЦТР.
- Компьютерная периметрия. При длительном повышении офтальмотонуса развиваются типичные глаукоматозные изменения поля зрения. Первыми появляются дугообразные скотомы, которые в последующем становятся множественными. Зрительное поле постепенно сужается с назальной стороны.
- Офтальмоскопия. Отклонения со стороны диска зрительного нерва представлены перипапилярной атрофией, истончением нейроретинального пояска с височной и нижней стороны. Толщина слоя нервных волокон сетчатки снижена преимущественно в нижнем квадранте.
Дифференциальная диагностика проводится с врожденной глаукомой, аномалией Петерса и гипоплазией радужки. У пациентов с врожденной глаукомой не удается визуализировать задний эмбриотоксон, что характерно для синдрома Аксенфельда-Ригера. При аномалии Петераса наблюдается центральный дисгенез роговицы и отсутствуют признаки гипоплазии радужки. При развитии симптомов болезни показана консультация офтальмолога и генетика.
Лечение синдрома Аксенфельда-Ригера
Лечение выполняется при наличии признаков ухудшения зрительных функций, повышения офтальмотонуса и фотофобии. В случае чрезмерной чувствительности к свету из-за патологических изменений радужки пациенту рекомендуют использовать контактные линзы. Альтернативой является только хирургическое вмешательство, направленное на коррекцию дефекта радужной оболочки.
Консервативная терапия
Медикаментозное лечение назначается при повышении внутриглазного давления. Наиболее целесообразно применение препаратов из группы бета-блокаторов, альфа-агонистов и ингибиторов карбоангидразы (КАГ), т. к. они способствуют снижению продукции внутриглазной жидкости. Необходимо отметить, что альфа-агонисты должны с осторожностью использоваться в детском возрасте из-за возможного угнетения функций центральной нервной системы.
Нейропротекторная терапия сводится к назначению препаратов, содержащих в своем составе ресвератрол, пирацетам и тиотриазолин. Средняя продолжительность курса – 10-14 дней. Доказана эффективность применения кортексина. Парабульбарно вводится эмоксипин. В комплекс лечения следует включить витамины В1, В6. Важно отметить, что цель терапии – предупредить прогрессирование глаукоматозных изменений.
Хирургическое лечение
Оперативное вмешательство показано при отсутствии эффекта от консервативной терапии, когда применение комбинированных препаратов не позволяет снизить ВГД до референтных значений. При синдроме Аксенфельда-Ригера выполняется трабекулоэктомия с дополнительным использованием антиметаболитов. Если пациент предъявляет жалобы на светобоязнь при гипоплазии радужки, рекомендовано проведение иридопластики.
Прогноз и профилактика
При нормальном офтальмотонусе прогноз у больных синдромом Аксенфельда-Ригера благоприятный. В таком случае, пациентам рекомендовано проходить плановые осмотры у офтальмолога 2 раза в год с обязательным измерением ВГД, проведением биомикроскопии и офтальмоскопии. В свою очередь, повышение ВГД ведет к развитию вторичной глаукомы, что в тяжелых случаях влечет за собой возникновение фтизиса глазного яблока. Специфическая профилактика не разработана. Неспецифические превентивные меры направлены на проведение медико-генетического консультирования, с целью предупреждения наследственных патологий у ребенка.
Источник