Синдром мышечной дистрофии у детей

Мышечная дистрофия у детей относится к заболеваниям наследственного характера. Происходит нарушение функции волокон. Данная патология может передаваться по наследству. Дистрофия мышц является неизлечимой болезнью. Только поддерживающая терапия может значительно улучшить состояние ребенка. Терапия заключается в том что, назначаются физиотерапевтические процедуры.
Разновидности мышечных дистрофий
Если изучить все виды дистрофии, то их существует огромное множество. Но все они встречаются довольно редко. Наблюдается четыре типа дистрофий:
- Миопатия псевдогипертрофического происхождения;
- Болезнь Беккера;
- Миотония врожденного генеза;
- Дистрофия мышц плеча и дегенерация газового участка.
Самым распространенным среди всех дистрофий является миопатия псевдогипертрофического происхождения. Часто встречается у мальчиков, у девочек такая патология не диагностируется. По статистическим данным встречается у каждого трехтысячного ребенка. Первые признаки заболевания появляются в раннем детстве. Далее происходит прогрессирование снижения функции мышечных волокон, что приводит к падению активности.
Что касается Болезни Беккера, то встречается реже, чем предыдущая патология. Клинические проявления более скудные, их поначалу даже сложно диагностировать. Но, так или иначе, ребенок становится инвалидом.
Если диагностирована миотония дистрофического или врожденного происхождения, то в первую очередь ребенку тяжело дышать – это является основополагающим симптомом. После чего, сразу начинают ослабевать все группы мышц больного. Одинаково болеют как девочки, так и мальчики.
Среди всех дистрофий, самым редко встречающимся видом является поражение мышц плеча и газового пояса. Очень тяжелая патология, ухудшается качество жизни малыша.
Причины
Если мальчик заболевает миодистрофией, то исход крайне неблагоприятный. Такие больные живут максимум до 22-х лет. Если же у ребенка диагностирована болезнь Беккера, то исходом является инвалидность. Если проходит 20 лет с начала заболевания, то резко ухудшается активность человека, вплоть до прикованности к креслу.
Что касается миотонии врожденного генеза, то такие детки долго не живут. Но были случаи, когда новорожденные переживали первые сутки, далее могли прожить еще лет 15, но не более.
Абсолютно все разновидности патологии возникают из-за некоторых сбоев в генетической цепи. Если глубоко вдаться в подробности, нарушается структура на Х-хромосоме. Данная единица отвечает за выработку такого белка, как дистрофин. Он необходим для формирования нормальной функции мышечной ткани. Если происходит сбой в этом белке, то наступает дисфункция волокон и всего связочного аппарата организма.
Женский пол при данном заболевании является «переносчиком» патологического гена. Очень редко болеют девочки. Это связано с тем, что женский пол имеет две Х- хромосомы. Исходя из этого, происходит компенсация со второй Х-хромосомы.
Как только передается дефектный ген плоду мужского рода, то мальчик начинает заболевать. Это потому, что у мужского пола одна Х-хромосома. Поэтому компенсации от второй хромосомы не получится никогда.
Если сыновья являются непосредственными носителями патологического гена, то шансы передать по наследству становится около 50%. И около 50% всех девочек являются носителями мышечной дистрофии. Наблюдались казуистические случаи, когда ребенок заболевал, но в роду данной патологии не наблюдалось.
Диагностика
Выявить у детей раннего возраста заболевание не представляет никакой сложности. Достаточно изучить анамнез больного ребенка и провести клиническое обследование. Для точности доктор берет кровь больного и изучает ее в лабораторных условиях. Если наблюдается повышенное количество в крови креатинфосфокиназы, то можно подозревать, что ребенок болен. В нормальном состоянии этот фермент находится в мышечных волокнах пациента.
Также для проведения диагностики применяют:
- Электромиографию (с точностью выявляет активность электрического потенциала мышечной ткани);
- Эхокардиограмму ( для исключения сердечной патологии, ведь сердце – это мышца);
- Биопсию мышечных волокон.
Биопсия берется у ребенка для того, чтобы изучить структурные изменения в волокнах. Это могут быть снижение коллагена или наличие избыточного отложение тканей жирового происхождения.
Лечение
В нынешнее время полностью купировать заболевание невозможно. Не существует каких-либо лекарственных препаратов или иных процедур, чтобы восстановить пораженные участки волокон.
Лечебная терапия данного патологического процесса направлена на приостановление прогрессирования деструкции. Для этого назначается:
- Витаминотерапия;
- Препараты АТФ;
- Кортикостероиды;
- Лечебная физкультура;
- Предупреждение развития сколиоза, а также контрактуры ног.
Если выполнять данные пункты, то можно притормозить развитие заболевания. Проводить лечение нужно исключительно после рекомендации врача. Если не соблюдать все настояния специалиста или вовсе не лечить ребенка, то может настигнуть летальный исход.
- Советуем почитать про: виды дистрофии у детей
Профилактика
Для того чтобы предупредить данное заболевание у будущего потомства, существуют некоторые рекомендации. К ним относится:
- Если мать планирует забеременеть, то необходимо провести лабораторное исследование на присутствие в организме генов патологического происхождения. Также нужно тщательно изучить генеалогическое древо, чтобы исключить мышечную дистрофию.
- Обследовать отца на наличие патологических генов. Ведь это также играет важную роль для предотвращения рождения больного ребенка.
- Строгое соблюдение всех профилактических мер в случае осложнений у пациента.
Если соблюдать профилактические пункты, то можно исключить появление детей с данным заболеванием.
Оцените статью: 86 Пожалуйста оцените статью
Сейчас на статью оставлено число отзывов: 86 , средняя оценка: 4,05 из 5
Загрузка…
Источник
Врожденная мышечная дистрофия у детей. Диагностика и лечение
Название врожденная мышечная дистрофия терминологически неверно, так как все мышечные дистрофии генетически детерминированы. Этот термин применяется для описания нескольких различных заболеваний со сходными признаками, включающими тяжелое поражение при рождении обычно с последующим неожиданно доброкачественным клиническим течением. Как правило, эти заболевания наследуются по аутосомно-рецессивному типу.
При рождении часто наблюдаются контрактуры или артрогрипоз и диффузная мышечная гипотония. Мышечная масса слабо развита на туловище и конечностях. Ребенок плохо удерживает голову. Возможно умеренное поражение мимических мышц, однако такие симптомы, как офтальмоплегия, слабость глоточных мышц и слабое сосание, нехарактерны. У небольшой части пациентов бывает тяжелая дисфагия, требующая регулярного зондового питания или наложения гастростомы. Сухожильные рефлексы могут быть снижены или отсутствовать. Артрогрипоз характерен для всех форм врожденной мышечной дистрофии.
Одна из форм врожденной мышечной дистрофии — болезнь Фукуямы — представляет собой вторую по распространенности мышечную дистрофию в Японии (после дистрофии Дюшенна). Это заболевание также встречается у детей в Дании, Германии, Скандинавии и Турции. При болезни Фукуямы поражение скелетной мускулатуры обычно сопровождается тяжелой кардиомиопатией и пороком развития головного мозга. В клинической картине выражены симптомы поражения этих органов: кардиомегалия, сердечная недостаточность, умственная отсталость, судороги, микроцефалия и дефицит массы тела.
Аномальный ген при врожденной мышечной дистрофии Фукуямы картирован в локусе 8q31-33 у японских пациентов.
Признаки поражения нервной системы могут сопровождать и другие формы врожденной мышечной дистрофии кроме болезни Фукуямы. Наибольшей изменчивостью отличается психический и неврологический статус пациентов; отсутствие неврологических нарушений и нормальный интеллект не позволяют исключить это заболевание, если имеются характерные клинические проявления, указывающие на эту форму миопатии. Пороки развития головного мозга могут варьировать от тяжелой дисплазии (голопрозэнцефалия, лиссэнцефалия) до менее распространенных поражений (агенезия мозолистого тела, локальная гетеротопия коры большого мозга и подкоркового белого вещества, гипоплазия мозжечка). Врожденная мышечная дистрофия постоянно сочетается с дисгенезиями мозга при синдроме Уолкера-Варбурга и болезни Сантавоури (болезни мышц-глаз-головного мозга).

Патоморфологические исследования выявляют аномалии миграции нейробластов в коре большого мозга, мозжечке и стволе мозга. Другая изолированная форма врожденной мышечной дистрофии характеризуется микроцефалией и умственной отсталостью.
Лабораторные исследования при врожденной мышечной дистрофии у детей. Уровень КФК в крови обычно умеренно повышен и достигает от нескольких сотен до многих тысяч ЕД/л. В некоторых случаях содержание КФК в крови может быть на верхней границе нормы. ЭМГ выявляет неспецифические миопатические изменения. При всех формах врожденной мышечной дистрофии в схему обследования необходимо включать оценку функции сердечной деятельности и методы нейро-визуализации мозга. Мышечная биопсия необходима для подтверждения диагноза.
Диагностика при врожденной мышечной дистрофии у детей. Мышечная биопсия имеет диагностическое значение как в неонатальном периоде, так и в более старшем возрасте. Определяется избыточная пролиферация коллагена эндомизия. Уже при рождении отдельные мышечные волокна окутаны соединительной тканью, при этом волокна принимают округлую форму в поперечном сечении и как бы окружены жесткой муфтой, что особенно заметно при их сокращении. Соединительная ткань перимизия и жировая ткань избыточно развиты, пучковая организация мышцы может быть нарушена вследствие фиброза. В культурах тканей внутримышечных фибробластов повышен синтез коллагена, при этом его структура не изменена.
Мышечные волокна варьируют в диаметре, могут выявляться центральные ядра, расщепление миофибрилл и другие изменения цитоархитектоники. Встречаются разрозненные волокна с дегенеративными и регенеративными изменениями. Отсутствуют воспалительные изменения или аномальные включения. Иммуноцитохимический метод выявляет отсутствие мерозина (а2-цепи ламинина) в области сарколеммы примерно в 50 % случаев и нормальное содержание мерозина у остальных пациентов. Мерозин — белок, связывающий сарколемму мышечных волокон с базальной пластинкой или основной мембраной. Дефект мерозина обусловлен мутацией гена LAMA2, расположенной в локусе 6q22-q23. Наличие или отсутствие мерозина не всегда коррелирует с тяжестью миопатии и не всегда позволяет прогнозировать течение заболевания.
Однако у пациентов с дефицитом мерозина имеется тенденция к более тяжелому поражению головного мозга и более тяжелым проявлениям миопатии. В некоторых случаях выявляется вторичное уменьшение количества адхалена (а-дистрогликана).
Лечение. Проводится поддерживающая терапия.
— Также рекомендуем «Эндокринные миопатии у детей. Тиреоидная миопатия»
Оглавление темы «Нервно-мышечные болезни у детей»:
- Конечностно-поясная мышечная дистрофия. Диагностика
- Плече-лопаточно-лицевая миопатия — болезнь Ландузи-Дежерина у детей. Диагностика и лечение
- Врожденная мышечная дистрофия у детей. Диагностика и лечение
- Эндокринные миопатии у детей. Тиреоидная миопатия
- Периодический паралич у детей. Калийзависимые параличи
- Злокачественная гипертермия у детей. Причины
- Гликогенозы у детей: болезни Гирке, Форбса-Кори, Андерсена, Мак-Ардла, Таруи
- Митохондриальные миопатии у детей. Причины и диагностика
- Миопатии с накоплением липидов. Дефицит карнитина у детей
- Миопатии с дефицитом витамина Е. Миастения Gravis у детей
Источник
Прогрессивная мышечная дистрофия. Формы и признаки мышечной дистрофии у детей
Под этим названием объединяется группа наследственных хронических первичных заболеваний поперечнополосатой мускулатуры, имеющих сходные клинико-морфологические проявления. Предполагается, что в основе патогенеза лежит ферментопатия, однако ферментный дефект не выяснен.
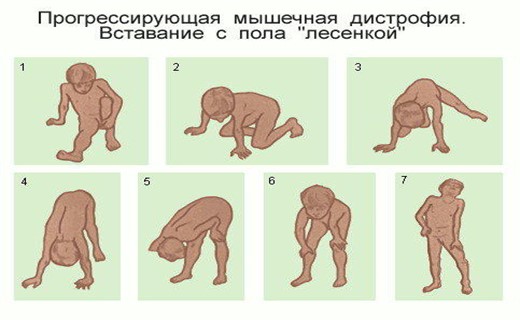
Ведущим клиническим признаком является медленно нарастающая большей частью симметричная атрофия мышц с снижением их силы пропорционально степени поражения. В зависимости от типа наследования, возраста больных, течения и локализации процесса выделяют три основные формы прогрессивной мышечной дистрофии.
Ранняя форма (Дюшенна) передается по рецессивному типу наследования, связанному с Х-хромосомой. Начинается в возрасте до 3 лет. Болеют почти исключительно мальчики. Сначала возникают изменения мышц тазового пояса и нижних конечностей, затем — плечевого пояса, позднее вовлекается другие группы мышц. В большинстве случаев во втором Десятилетии жизни наступает смерть от интеркуррентного заболевания органов дыхания.
Юношеская форма (Эрба) характеризуется аутосомно-доминантным типом наследования. Чаще начинается в период полового созревания и протекает медленно. Изменение проявляются прежде всего в области плечевого пояса, иногда на лице (веки, губы; миопатическое лицо — гладкий лоб, недостаточное смыкание глаз, толстые губы), в дальнейшем распространяются на мышцы туловища и таза.
Нередко отдельно описывают плече-лопаточно-лицевую форму («Пандузи — Дежерина), а к форме Эрба относят изменения, начинающиеся с мышц тазового, реже плечевого пояса с распространением на мышцы туловища и конечностей.
Форма с аутосомно-рецессивным путем наследования (Лейдена) имеет общие признаки с двумя предыдущими формами. Начинается, как правило, несколько раньше и протекает быстрее, чем юношеская. При возникновении в раннем возрасте болезнь быстро прогрессирует и заканчивается смертью.
В ряде случаев клинические симптомы заболевания с предположительным аутосомно-рецессивным типом наследования проявляются с рождения (врожденная мышечная дистрофия). Изменения скелетной мускулатуры при этом не отличаются от наблюдаемых при прогрессивной мышечной дистрофии.
Макроскопически измененные мышцы часто напоминают рыбье мясо вследствие обеднения их миоглобином. Объем мышц уменьшен, иногда не изменен и даже увеличен за счет вакатного разрастания волокнистой соединительной ткани и жировой клетчатки, что особенно характерно для ранней формы Дюшенна.
Макроскопически типичны вариабельность диаметра мышечных волокон (от 7—15 до 100—250 мкм), наличие наряду с истонченными нормальных и отдельных гипертрофированных волокон. В волокнах наблюдаются атрофические, дистрофические (уменьшение содержания гликогена и увеличение содержания липидов, вакуолизация, гиалиноз, ослабление поперечной исчерчеиности) и некробиотические (глыбчатый распад) изменения. Отмечается продольное расщепление волокон, увеличение числа ядер и центральное их расположение.
Активность окислительных и гликолитических ферментов долгое время остается высокой, вместе с тем дифференцировка волокон на первый (красные) и второй (белые) типы, имеющие разные ферментативные свойства, становится менее четкой. При количественной оценке В. В. Семенова-Тян-Шанская и Л. А. Сайкова (1977) обнаружили снижение активности сукцинатдегидрогеназы и лактатдегидрогеназы. При тяжелом течении миопатии встречаются лишь небольшие участки мышечных волокон среди обширных разрастаний соединительной и жировой ткани.
При электронно-микроскопическом исследовании на ранних стадиях обнаруживают изменения миофиорилл в виде деструкции Z-полос, позднее — распад сократительных элементов. В случаях с тяжелым течением заболевания видны также деструктивные изменения цитоплазматической сети и митохондрий, увеличение количества лизосом.
Дифференциальную диагностику следует проводить с неврогенными формами мышечной атрофии, прежде всего с прогрессивной спиналыюй мышечной атрофией (спиналыюй амиотрофией) детского возраста (болезнь Верднига — Гоффманна), развивающейся в первые годы жизни в связи с повреждением передних рогов спинного мозга.
При этой болезни наблюдаются дистрофические изменения нервных клеток передних рогов, уменьшение их числа, иногда полное исчезновение, разрастание глиальных элементов, демиелинизация передних корешков и периферических нервов. Сначала поражаются мышцы тазового пояса, нижних конечностей, затем — плечевого пояса и другие. В отличие от прогрессивной мышечной дистрофии атрофированные мышечные волокна образуют группы (пучковая атрофия), а не располагаются вперемежку с сохранившими свою толщину или гипертрофированными волокнами.
Ранним признаком неврогенных атрофий считается потеря «шахматного» распределения волокон первого и второго типов с исчезновением различий между ними. Если болезнь начинается в школьном пли пубертатном возрасте, говорят о юношеской форме спиналыюй мышечной атрофии (болезнь Кугельбсрга — Веландера). При врожденной миатонии (болезнь Оппенгейма) изменения мышц сходны с изменениями, наблюдающимися при прогрессивной мышечной дистрофии. Вместе с тем в большинстве случаев выявляются изменения нервных клеток передних рогов спинного мозга, что сближает миатонию Оппенгейма со сшшальной мышечной атрофией Верднига — Гоффманна.
— Также рекомендуем «Врожденная миотония — болезнь Томсена. Миотоническая дистрофия, синдром Краббе»
Оглавление темы «Врожденные заболевания хрящей, костей и мышц детей»:
1. Спазмофилия при рахите. Витамин-D-резистентный рахит у детей
2. Врожденная идиопатическая гиперкальциемия и гипофосфатазия. Несовершенный остеогенез
3. Признаки несовершенного остеогенеза. Остеопетроз — мраморная болезнь,болезнь Альберс — Шенберга/a>
4. Диагностика остеопетроза. Хондродистрофия у детей
5. Хондроматоз костей — болезнь Олье. Костно-хрящевые экзостозы новорожденных
6. Фиброзная дисплазия. Признаки фиброзной дисплазии костей
7. Прогрессивная мышечная дистрофия. Формы и признаки мышечной дистрофии у детей
8. Врожденная миотония — болезнь Томсена. Миотоническая дистрофия, синдром Краббе
9. Немалиновая миопатия. Митохондриальная миопатия
10. Врожденные болезни иммунной системы. Возрастная инволюция тимуса
Источник
Группа наследственных заболеваний мышц, которые характеризуются медленной прогрессирующей дегенерацией мышечных волокон — это мышечная дистрофия. Этот термин охватывает ряд наследственных заболеваний, характеризующихся слабостью и атрофией мышц. Она проявляется прогрессирующей слабостью и дегенерацией мышц. В этой статье мы расскажем вам про основные причины заболевания и способы его диагностики у ребенка.
Причины
Для мальчиков с псевдогипертрофической миопатией прогноз неблагоприятный, они обычно доживают только до 20 лет.
Большинство мальчиков в конце концов становятся тяжелыми инвалидами. Через 25 лет после начала болезни они обычно оказываются прикованными к инвалидному креслу.
Врожденная миотония может быть смертельной, хотя дети, пережившие свой первый день рождения, чаще всего доживают до совершеннолетия.
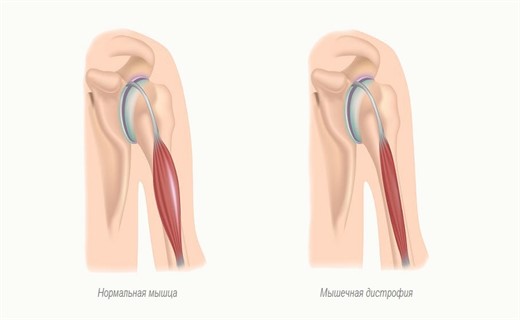
Все виды обусловлены генетическим дефектом. Причиной мышечной дистрофии и псевдогипертрофической миопатии является дефект структуры гена на Х-хромосоме (половая хромосома, имеющаяся у обоих полов). Этот ген ответственен за образование дистрофина — белка, необходимого для здоровых мышц. Без дистрофина нарушается нормальная структура мышечных волокон и пораженные мышцы слабеют.
Половая хромосома — причина мышечной дистрофии
1.
Девочки могут быть носителями дефектного гена, но обычно не страдают болезнью, т. к. имеют две Х-хромосомы, и нормальный вариант гена на второй Х-хромосоме компенсирует дефект.
2.
Если дефектный ген передается потомству мужского пола, то мышечная дистрофия у детей появляется только у мальчиков, т. к. у них только одна Х-хромосома и, следовательно, нет нормального варианта гена, способного компенсировать дефект.
У сыновей носителей дефектного гена, вероятность унаследовать его и заболеть составляет 50% их дочери в 50% случаев становятся носителями дефектного гена. Хотя эта генная аномалия часто наследуется, иногда дистрофия спонтанно возникает у мальчика, в семье которого никто и никогда не болел ей.
Мышечная дистрофия плечевого и тазового пояса — это аутосомное рецессивное расстройство: ребенок заболевает тогда, когда оба родителя являются носителями дефектного гена.
Классификация
Существует много видов заболевания и все они достаточно редки. Ниже описаны четыре вида болезни:
- Псевдогипертрофическая миопатия — наиболее распространенный вид заболевания, поражает только мальчиков и диагностируется примерно у одного ребенка из трех тысяч. Возникает в детстве и ведет к прогрессирующему снижению подвижности.
- Болезнь Беккера встречается реже, чем псевдогипертрофическая миопатия, протекает с более слабыми симптомами, но также приводит к прогрессирующей инвалидности.
- Врожденная и дистрофическая миотонии поражают как девочек, так и мальчиков. У больных отмечаются затруднения с дыханием и слабый мышечный тонус.
- Мышечная дистрофия плечевого и газового пояса поражает девочек и мальчиков и затрагивает мышцы плеч и газа.
Диагностика
Болезнь неизлечима, хотя физиотерапия может значительно облегчить состояние пациента. Для диагностики мышечной дистрофии часто достаточно выяснить симптомы и провести обследование.
Врач берет кровь для анализа, т. к. при заболевании в крови заметно повышен уровень креатипфосфокипазы (фермент, в норме присутствующий в здоровых мышцах).
- Электромиография позволяет определить потенциал электрической активности в мышце в случае дистрофии эти потенциалы не соответствуют норме;
- У больного ребенка мышечная биопсия (маленький образец ткани, взятый на анализ) показывает характерные изменения в структуре мышцы. При диагностике выявляются поврежденные мышечные волокна и аномальное отложение жировой ткани;
- Может быть также сделана эхокардиограмма, чтобы определить, затронута ли сердечная мышца.
Теперь вы знаете основные способы диагностики и причины мышечной дистрофии у детей. Здоровья вашему ребенку!
Источник