Синдром марфана у девочек фото

Интервью: Ксения Акиньшина
О СУЩЕСТВОВАНИИ ГЕНЕТИЧЕСКИХ ЗАБОЛЕВАНИЙ СЛЫШАЛИ ВСЕ, но мало кто представляет, как их много. Не все выявляются при рождении: например о синдроме Марфана становится известно гораздо позже. Это генетическое заболевание, при котором нарушается синтез белка фибриллина — он отвечает за эластичность и сократимость соединительной ткани. Поражаются многие системы и ткани организма: сосуды и сердце, кости и суставы, глаза и лёгкие. Люди с этим заболеванием обычно высокие, с длинными руками, ногами, кистями и стопами; их вытянутые пальцы в медицинских учебниках принято называть «паучьими». Лечение и прогноз напрямую зависят от степени тяжести: например, если поражена аорта — самый большой сосуд в организме человека — состояние становится угрожающим жизни. Мы поговорили со Светланой Х. о том, как она живёт с синдромом Марфана.
Мне тридцать лет, а о диагнозе стало известно, когда мне было шесть. Я быстро росла, и на очередном плановом осмотре врач услышал шумы в сердце; после этого был приём у генетика и предположительный диагноз — синдром Марфана; в статусе «предположительного» он оставался многие годы. Болезнь тогда была плохо изучена, и единственными рекомендациями были наблюдение кардиолога и запрет на физические нагрузки. Потом, правда, стало понятно, что необходима лечебная физкультура, и я пошла на плавание. Получалось хорошо: я занималась в школе олимпийского резерва и в девять лет меня даже пригласили в профессиональную команду. Кардиолог и родители, правда, были против увеличения нагрузок — расстроившись, я бросила плавание вообще.
По мере взросления проблемы с сердцем ухудшались: я росла, а вместе со мной растягивалась аорта — самый большой сосуд организма. Уже в детском возрасте все параметры аорты превышали нормальные значения даже для взрослого человека. Проблемы с клапанами сердца тоже шли по нарастающей. В десять лет мне довелось побывать на консилиуме врачей — в то время это было редкостью, да ещё в бесплатной медицине, то есть случай был тяжёлым. Решался вопрос кардиохирургии, но прямых показаний к операции не было — просто «вялотекущее ухудшение», да и «неизвестно, как поведут себя ткани после операции, может быть, будут расползаться». Генетически подтвердить или опровергнуть диагноз тогда не предлагали — то ли врачи были не в курсе такой возможности, то ли её тогда просто не существовало.
Течение болезни бывает очень тяжёлым, и к лечению нужно подходить ответственно, поскольку на кону не только качество жизни, но и она сама: по статистике 90–95 % пациентов не дотягивают до 40–50 лет
В общем, единственными обследованиями были ЭКГ, ЭХО, холтеровское мониторирование (круглосуточная ЭКГ в условиях обычной повседневной активности, когда датчики ЭКГ приклеивают к телу. — Прим. ред.), посещения кардиолога, поддерживающая терапия и совет «не болеть». Следовать ему у меня не очень-то получалось. В тринадцать лет, пролежав дома две недели с температурой под сорок, я всё же попала в инфекционную больницу — и в приёмном отделении врач, увидев моё горло, побледнела и объявила моей маме, что у меня дифтерия. Мама расплакалась, а меня перевели в отделение и поместили в палату с выздоравливающими от гриппа — «чудесное» решение. Хорошо, что диагноз не подтвердился и дифтерии у меня всё же не было. Тем не менее любая болезнь, тем более поражающая дыхательные пути, негативно отражается на сердце, что в моём случае просто опасно.
Всю жизнь я прожила с диагнозом «дисплазия соединительной ткани», а синдром Марфана был лишь фенотипом — это значит, что у меня были проявления синдрома, но он не был подтверждён генетически. По мнению некоторых врачей, особенно тех, кто видел меня впервые, он отсутствовал вовсе — ведь полного набора классических симптомов не было. У меня всё более или менее нормально с костями и состоянием позвоночника, на месте хрусталик глаза; болезнь можно заподозрить только из-за патологий сердечно-сосудистой системы, высокого роста, арахнодактилии (те самые «паучьи пальцы») и повышенной эластичности. Я и сейчас, рассказывая врачам об анамнезе, упоминаю дисплазию чаще, чем синдром Марфана — иначе они просто вежливо кивают и пропускают мимо ушей.
Поскольку меня не беспокоила боль, я жила как обычный подросток. За моё сердце переживала мама, но неполная осведомлённость помогла пройти этот путь гораздо легче. Если бы тогда она была в курсе всех сюрпризов, которые может преподнести болезнь, не знаю, как справлялась бы с этим. Я даже рада, что и сейчас она знает не больше прежнего — теперь уже я думаю о её сердце. Реально же подтвердить диагноз удалось только на тридцатом году жизни: я самостоятельно сдала анализ на мутации генов в рамках так называемой панели заболеваний соединительной ткани. С его помощью выявили мутацию, характерную для синдрома Марфана, однако не в «горячих точках» — возможно, поэтому я и не типичный представитель болезни.
Синдром Марфана — это генетическое заболевание, но оно не обязано проявляться у кого-то из родни. У моих родителей, например, нет никаких проявлений, ни внешне, ни «внутренне». В основе заболевания лежат мутации в гене, отвечающем за синтез фибриллина — важнейшего структурного белка межклеточного матрикса, придающего эластичность и сократимость соединительной ткани. При нём сильнее всего страдают органы с наибольшей «концентрацией» соединительной ткани: сердце, глаза, спина, связки. Синдром не лечится, и любая терапия направлена на конкретные пострадавшие органы — например, я должна пить курсами таблетки, помогающие сердечно-сосудистой системе. Течение болезни бывает очень тяжёлым, и к лечению нужно подходить ответственно, поскольку на кону не только качество жизни, но и она сама: по статистике 90–95 % пациентов не дотягивают до 40–50 лет.
Ограничения в первую очередь касаются физических нагрузок. Нельзя заниматься профессиональным спортом, хотя по иронии судьбы данных у меня как раз предостаточно — например, высокий рост и колоссальная гибкость (до сих пор могу закинуть ногу за голову или сесть в позу лотоса с разбега). Людям с синдромом Марфана показан разумный спорт без резких движений, например плавание. В путешествиях моя аптечка не больше, чем у обычного человека, никакой специфики — но тут дело скорее не в факте синдрома, а в степени его проявления.
Из-за «кукольного театра», как назвали ширму между мной и оперирующими врачами, я слышала, как акушер постоянно повторяла ассистентам: «Не рвите ткани, не рвите ткани!»
Со всей серьёзностью проблемы я столкнулась, когда мы с мужем захотели ребёнка. Генетик приговорил к суррогатному материнству или возможной беременности после кардиохирургии, к которой не было прямых показаний. Мне объясняли, что функции сердца достаточно для моего собственного жизнеобеспечения, но при беременности нагрузка удваивается. Я же настаивала, что при относительно лёгкой степени поражения сердца и сосудов нормальное течение беременности возможно. Я побывала во всех кардиоцентрах Москвы, пролила море слёз и всё же получила разрешение, при условии постоянного наблюдения кардиолога. Так, через два месяца я пришла вставать на учёт в кардиологический перинатальный центр, который согласился меня вести.
Конечно, решиться на беременность было очень страшно, особенно после неоднозначного прогноза врачей — моя аорта находится пока на докритическом уровне. Надеюсь, там она и останется — до критического лишь несколько миллиметров. Надежду вселила врач-гинеколог, посвятившая полжизни изучению дисплазии соединительной ткани. После разговора с ней я поняла, что если я не попробую выносить ребёнка вопреки своему желанию, то буду жалеть всю жизнь. Где-то глубоко внутри я взяла на себя этот риск и не жалею.
Беременность прошла отлично, а дочь родилась через плановое кесарево сечение немного раньше срока. Врачи безумно боялись, что аорта «рванёт» от максимальных нагрузок и я умру прямо в роддоме, где была самой «тяжёлой» роженицей. Дочь — моя победа, и мы назвали её Викторией. Из-за «кукольного театра», как назвали ширму между мной и оперирующими врачами, я слышала, как акушер постоянно повторяла ассистентам: «Не рвите ткани, не рвите ткани!» — но в тот момент я была готова ко всему, лишь бы с ребёнком всё было в порядке. Операция длилась в два раза дольше обычного, врач была мокрой, будто на неё вылили ведро воды. Я пару раз почти теряла сознание, в чувства приводил анестезиолог, а потом, уже в реанимации, я узнала, что потеряла почти литр крови. Состояние моей сердечно-сосудистой системы осталось без изменений, то есть таким же, как до беременности.
Оглядываясь на свою юность, я понимаю, что мне в какой-то степени повезло, если можно так выразиться. Да, меня не обошли высокий рост, пластинка на зубах, одно плечо выше другого — конечно, я выделялась из толпы, были насмешки среди сверстников и слёзы ночами в ванной комнате. Но такое было у многих, такова подростковая жизнь. После общения с мамами детей с более тяжёлым синдромом Марфана, я поняла, что моя жизнь могла бы быть гораздо труднее.
Свой оптимизм я взрастила сама, по крупинкам — при этом я заядлый параноик, что вообще присуще людям с синдромом Марфана. В интернете есть множество статей и информации, в которой очень легко запутаться и накрутить себя ещё сильнее. Специалистов же, которые разбираются в проблеме, единицы. Есть группы и форумы, где люди описывают свои симптомы, делятся опытом и даже «ставят» себе и другим диагнозы; многие хорошо изучили проблему и дадут в этой области фору некоторым докторам. Но большинство пишут о неизбежности, о моральной и физической боли, о доживании — поэтому я в этих группах не сижу, не хочу загонять себя в переживания ещё глубже. Конечно, я понимаю свои перспективы, но всегда ищу положительное в окружающем мире, стараюсь не зацикливаться на проблемах — иначе крайне сложно выбраться из разрастающейся паники. Конечно, не нужно закрывать глаза на свой диагноз, будто его нет — он есть, и очень опасен, но это не клеймо и не приговор.
О моей особенности знают лишь самые близкие, и многие люди из окружения задают вопросы о втором ребёнке. Но я не могу решиться на этот шаг, просто не имею права. Ещё до рождения дочери я взяла на себя колоссальную ответственность за неё и перед ней. Возможно, мне придётся столкнуться с кардиохирургией, и мне очень страшно. Мой организм с годами даёт о себе знать всё больше, количество визитов к врачам ежегодно растёт — но это не повод сидеть и считать оставшиеся дни. Мне бывает очень сложно. Мысли о судьбе дочери, которой уже два года, и о продолжительности собственной жизни порой неделями не дают спать, но я делаю всё возможное, чтобы минимизировать проявления синдрома — ведь если я сдамся, лучше не станет. Нужно жить свою жизнь, а не проживать её.
Фотографии: spacedrone808 — stock.adobe.com (1, 2, 3)
Источник
Что это за болезнь?
Синдром Марфана – это наследственное заболевание, передающееся по аутосомно-доминантному типу и характеризуется поражением соединительной ткани и ее компонентов.
Болезнь Марфана вызывается мутированием гена, кодирующего фибриллин -1.
Люди с синдромом Марфана имеют удлиненные конечности, паукообразные пальцы и слабый (недоразвитый) подкожно-жировой слой и сверхгибкие суставы (см. фото ниже).
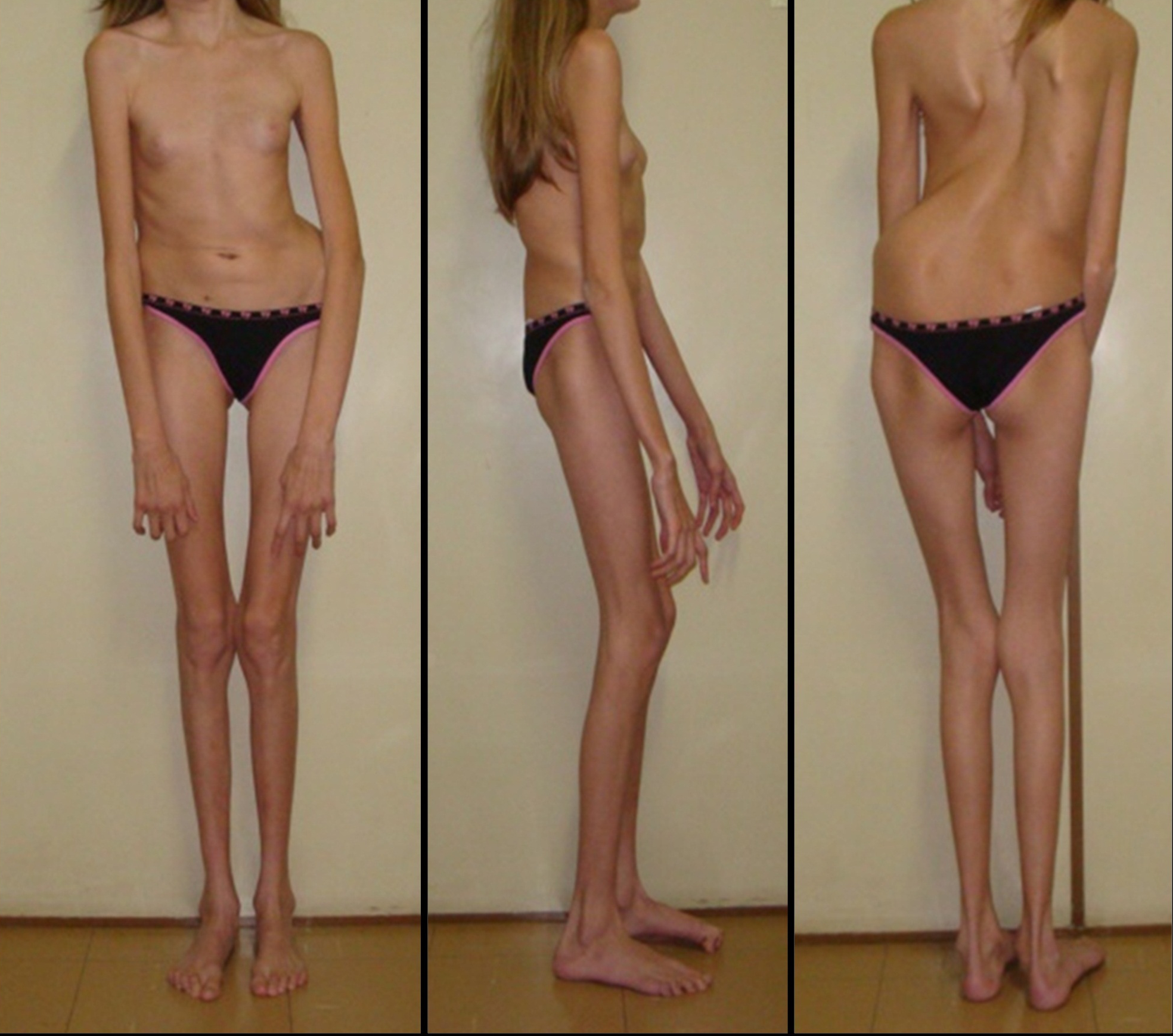
Кроме изменений костно-суставной системы, характерны изменения зрительного анализатора и сердечно-сосудистой системы. Также возможно поражение нервной, дыхательной и других систем.
Впервые описал данную патологию Вильямс, который заметил у своих брата и сестры – выпадение хрусталика, при этом они были очень высокими и имели гипермобильные суставы. Затем заметил Марфан, врач – невролог, у которого в течение 20 лет наблюдалась женщина с подобными симптомами, а затем еще 20 детей.
Причины возникновения
Болезнь Марфана у детей наследуется по аутосомно-доминантному типу (т. е передается от родителя к ребенку).
Также возможны мутации за счет воздействия на организм женщины факторов внешней среды (ионизирующее излучение, лучевая терапия, радиация).
Причины возникновения и механизм развития заболевания недостаточно изучены.
Особая роль отводится нарушению процессов обмена, в результате которых накапливается в коллагеновых и эластических волокнах большое количество мукополисахаридов.
Это ведет к тому, что соединительная ткань перерастягивается, легко подвергается механическому воздействию и приводит к развитию клинической симптоматики.
Классификация
Выделяют следующие формы болезни Марфана:
В зависимости от генной предрасположенности:
- семейная (патология передается от родителя к ребенку);
- спорадическая (патология вызвана внезапным мутированием в геноме).
В зависимости от проявлений клиники:
- стертая, когда признаки заболевания практически не проявляются и могут быть не замечены в течение всей жизни. Патологические изменения выявляются в одной или двух системах.
- выраженная, когда признаки заболевания касаются двух и более органов и систем (сердце, кости и суставы, легкие, кожа, глаза).
Симптомы болезни Марфана
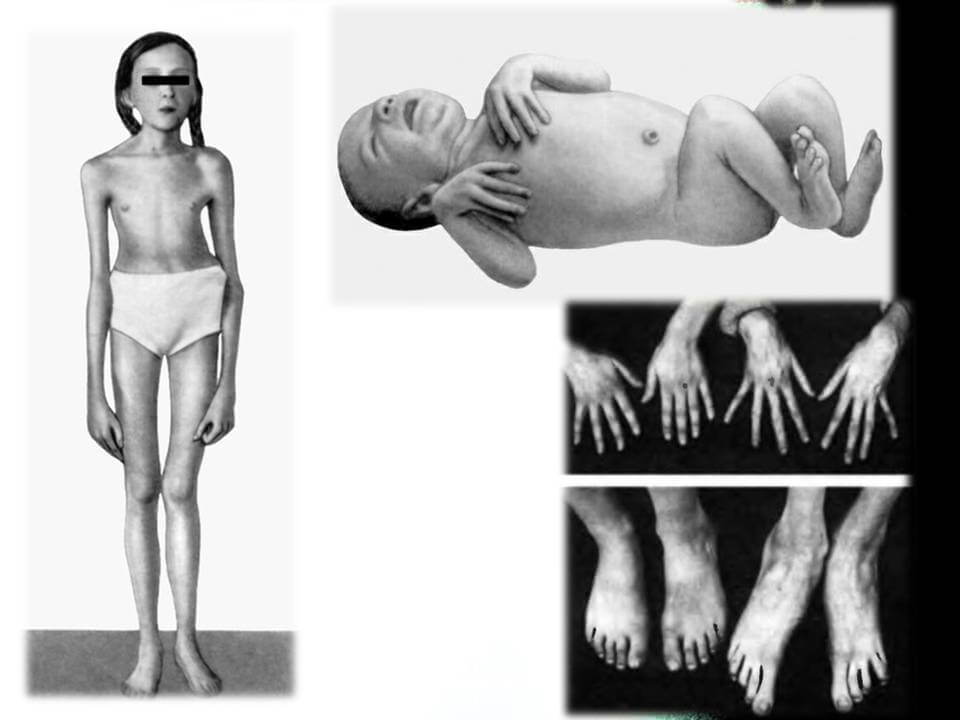
Синдромом Марфана у людей ведет к их выделению в обществе своим непропорциональным строением скелета. Для новорожденных на ранней стадии болезни, характерны длинные пальцы на руках, а к 7-9 годам у детей формируется развернутая клиническая картина.
У взрослых характерна различная симптоматика в зависимости от системы поражения:
- Нервная система: болезненность в поясничной области, головные боли,поражение симпатической и парасимпатической иннервации органов брюшной полости и малого таза (слабость кишечной стенки, недержание мочевого пузыря у ребенка). Также высок риск развития инсульта, субарахноидального кровоизлияния и разрыва аневризм головного мозга.
- Сердечно – сосудистая система: пороки сердца (сужение легочной артерии, пролабирование створок двустворчатого клапана, дилатационная кардиомиопатия, расширение границ сердца (аорты и всех ее отделов), дефекты МЖП и МЖЖ перегородок. У больных может развиваться нарушение ритма и проводимости в виде аритмий.
- Опорно-двигательный аппарат: телосложение астенической формы (дети худые), высокий рост у мужчин 190±10 см, у женщин 179±8 см, слаборазвитый подкожно-жировой слой, длинные пальцы (паукообразные), плоскостопие, череп и лицо вытянутые и узкие, недоразвитость скул, нарушение развития зубов и прикуса, вытянутая нижняя челюсть, готическое верхнее небо, гипермобильность суставов (смотрите на картинки выше). С возрастом ребенка может прогрессировать деформация позвоночного столба, с развитием сколиоза. Также может деформироваться грудная клетка, образуется вдавление – «грудь сапожника». Деформированный тазобедренный сустав нередко приводит к инвалидности при неоказанном своевременном лечении.
- Орган зрения: смещение хрусталика за счет слабого связочного аппарата) на ранней стадии, уплощение роговицы, развитие близорукости или дальнозоркости, спазм аккомодации, отслоение сетчатки.
- Кожа и мягкие ткани: перерастяжение кожи с образованием стрий атрофического характера. Они возникают внезапно, не связаны с колебание веса людей, беременностью и гормональным фоном. Кожа липкая, потная, с мраморным оттенком. Подкожно-жировой слой слабо развит, поэтому у больных наблюдаются грыжевые выпячивания в области передней брюшной стенки.
- Дыхательная система: развитие буллезной эмфиземы легких, проявляющейся кашлем, одышкой, развитием дыхательной недостаточности и спонтанного пневмоторакса.
Другие признаки:
- развитие опущения почек (нефроптоз);
- выпадение органов малого таза (опущение матки у женщин, или ее полное выпадение);
- варикоз вен нижних конечностей;
- запоры.
Диагностика
Диагностика основана на тщательном сборе анамнеза заболевания, выраженности клинической картины, данных осмотра, на результатах лабораторных и инструментальных методов исследований.
Сбор анамнеза включает в себя: наличие в семье данной патологии (родители, братья, сестры) или наличие факторов, провоцирующих мутацию в геноме человека.
К лабораторным методам относят: анализ генотипа ДНК с мутирующим геном, определение гликозаминогликанов в моче.
К инструментальным методам исследования относят:
- ЭКГ, служит для обнаружения патологии сосудов и сердца (ССС). Выявляют характерные нарушения ритма и проводимости в виде мерцательной аритмии, желудочковой экстрасистолии, развитие дилатационной гипертрофии миокарда левого желудочка.
- ЭхоКГ, также служит для обнаружения патологии ССС. Выявляют расширение аорты и ее структур, пролабирование двустворчатого клапана, увеличение размеров левой половины сердца.
- УЗИ сердца проводится для определения осложнений (расслаивающаяся аневризма).
- Рентген органов грудной клетки (изменения скелета, расширение полостей сердца, корней легких и др.)
- Компьютерная томография, магнитно-резонансно-ядерная томография позволяет выявить патологии костно-суставной, нервной системы, нарушение кровообращения в сосудах головного и спинного мозга.
Данные методы исследования служат для обнаружения критериев синдрома Марфана в различных органах и системах. Они играют самую важную роль для постановки и подтверждения диагноза, а в последующем, для определения тактики лечения.
Существуют следующие критерии диагностики синдрома Марфана:
| Система | Большие критерии | Малые критерии |
| Опорно- двигательный аппарат Должны быть: 4 больших критерия, либо 2 больших и 1 малый. |
|
|
| Орган зрения | Смещение хрусталика | Уплощенная роговица, близорукость, дальнозоркость, недоразвитие радужки и цилиарной мышцы глаз. |
| Сердечно-сосудистая система | Расширение аорты и ее структур | Пролабирование двустворчатого клапана, расширение клапана легочной артерии у лиц, не достигших 40 лет, отложение солей кальция на створках двустворчатого клапана, расслаивание аорты. |
| Дыхательная система | Отсутствуют | Внезапно развивающийся пневмоторакс (скопление воздуха в грудной клетке), верхушечные буллы. |
| Кожа | Отсутствуют | Повторное развитие грыжевых выпячиваний, атрофические стрии. |
| Нервная система | Расширения сосудов оболочек спинного мозга в поясничномкрестцовом отделе позвоночного столба. | Отсутствуют |
| Генетические изменения | Наличие данных критериев у родителей, детей, братьев, сестер, бабушек, дедушек. Наличие мутирующего гена, кодирующего фибриллин 1. | Отсутствуют |
Для постановки диагноза «Синдром Марфана» учитывается один признак из перечня больших критериев или малый критерий, характерный каждой из пораженной систем, кроме опорно-двигательного аппарата, (необходимо, как минимум, 4 критерия), а также наличие в семейном анамнезе больных с данной патологией.
Лечение синдрома Марфана
От синдрома Марфана полностью избавиться и устранить механизм его развития невозможно. Лечение базируется на улучшении общего состояния больного, устранении клинических проявлений и проведении профилактических мероприятий, препятствующих развитию осложнений.
Больным с данным синдромам рекомендовано ограничить физическую нагрузку до низкого уровня, или минимального. Риск появления патологии сердечно-сосудистой системы возрастает при средних и высоких физических нагрузках.
Следует обходить стороной и повседневные нагрузки, при которых возможно повышение внутригрудного давления, ведущему к развитию пневмоторакса (например, подъем тяжестей, подъем по этажам).
Больные с синдромом Марфана должны консультироваться у разных специалистов, в зависимости от клинически пораженных систем органов. Следует проходить медицинские осмотры каждые полгода в течение всей жизни.
Медикаментозное лечение
Медикаментозная терапия направлена на устранение клинической картины заболевания.
Со стороны сердечно-сосудистой системы рекомендованы β-адреноблокаторы (например: Анаприлин), которые снижают скорость распространения пульсовых волн при стремительно растущем расширении аорты и обратного тока крови на двустворчатом клапане или клапане аорты.
β-адреноблокаторы также оказывают положительный эффект при нарушении ритма и проводимости, в сочетании с сердечными гликозидами.
Но следует помнить о существующих противопоказаниях данных групп препаратов:
- хронический обструктивный бронхит;
- бронхиальная астма;
- снижение частоты сердечных сокращений;
- низкое артериальное давление.
Блокаторы каналов кальция используются при наличии противопоказаний к В-адреноблокаторам.
Хирургия
Хирургическое лечение проводится, если есть осложнения со стороны сердечно-сосудистой системы, с целью коррекции пораженных участков. Его проводят при пролабировании двустворчатого клапана и расслаивании аорты.
При этом осуществляется протезирование двустворчатого клапана.
У беременных с тяжелым течением болезни Марфана, роды разрешаются хирургическим путем.
Профилактика
С профилактической целью, во избежание развития инфекционных осложнений, образования тромбов и тромбоэмболии, назначаются антикоагулянты (гепарин), антибактериальная терапия и витаминотерапия.
- При синдроме Марфана с тяжелым поражением зрительного анализатора проводится хирургическая коррекция зрения, после которой пациенты должны носить очки или контактные линзы.
- Если возникают осложнения, проводят лазерную коррекцию глаукомы, катаракты, удаляют смещаемый хрусталик, заменяя его на искусственный.
- При функциональной дисфункции опорно-двигательного аппарата возникает необходимость стабилизирования позвоночника с помощью металлических пластин.
- При выраженной деформировании грудной клетки проводится торакопластика.
- При протрузии тазобедренных суставов производят внутреннее протезирование суставов.
Прогноз
Длительность жизни в среднем при синдроме Марфана составляет 30-45 лет.
Известно, что это многие знаменитые личности страдали данным синдромом. Это и Ганс Христиан Андерсен – датский писатель, автор знаменитой Русалочки; Авраам Линкольн – 16 президент США, Майкл Феллпс- известный пловец, многократный олимпийский чемпион. А также известные композиторы – Никколо Паганини, Сергей Рахманинов.
Люди с данной патологией должны тщательно следить за своим здоровьем, постоянно наблюдаться и консультироваться со своим лечащим врачом, избегать чрезмерных физических нагрузок.
По мимо медикаментозного лечения, необходимо проведение профилактических мероприятий с целью улучшения общего самочувствия, повышения иммунитета, соответствующий режим труда и отдыха.
Видеозаписи по теме
Источник