Синдром марфана кто живет с этим

Интервью: Ксения Акиньшина
О СУЩЕСТВОВАНИИ ГЕНЕТИЧЕСКИХ ЗАБОЛЕВАНИЙ СЛЫШАЛИ ВСЕ, но мало кто представляет, как их много. Не все выявляются при рождении: например о синдроме Марфана становится известно гораздо позже. Это генетическое заболевание, при котором нарушается синтез белка фибриллина — он отвечает за эластичность и сократимость соединительной ткани. Поражаются многие системы и ткани организма: сосуды и сердце, кости и суставы, глаза и лёгкие. Люди с этим заболеванием обычно высокие, с длинными руками, ногами, кистями и стопами; их вытянутые пальцы в медицинских учебниках принято называть «паучьими». Лечение и прогноз напрямую зависят от степени тяжести: например, если поражена аорта — самый большой сосуд в организме человека — состояние становится угрожающим жизни. Мы поговорили со Светланой Х. о том, как она живёт с синдромом Марфана.
Мне тридцать лет, а о диагнозе стало известно, когда мне было шесть. Я быстро росла, и на очередном плановом осмотре врач услышал шумы в сердце; после этого был приём у генетика и предположительный диагноз — синдром Марфана; в статусе «предположительного» он оставался многие годы. Болезнь тогда была плохо изучена, и единственными рекомендациями были наблюдение кардиолога и запрет на физические нагрузки. Потом, правда, стало понятно, что необходима лечебная физкультура, и я пошла на плавание. Получалось хорошо: я занималась в школе олимпийского резерва и в девять лет меня даже пригласили в профессиональную команду. Кардиолог и родители, правда, были против увеличения нагрузок — расстроившись, я бросила плавание вообще.
По мере взросления проблемы с сердцем ухудшались: я росла, а вместе со мной растягивалась аорта — самый большой сосуд организма. Уже в детском возрасте все параметры аорты превышали нормальные значения даже для взрослого человека. Проблемы с клапанами сердца тоже шли по нарастающей. В десять лет мне довелось побывать на консилиуме врачей — в то время это было редкостью, да ещё в бесплатной медицине, то есть случай был тяжёлым. Решался вопрос кардиохирургии, но прямых показаний к операции не было — просто «вялотекущее ухудшение», да и «неизвестно, как поведут себя ткани после операции, может быть, будут расползаться». Генетически подтвердить или опровергнуть диагноз тогда не предлагали — то ли врачи были не в курсе такой возможности, то ли её тогда просто не существовало.
Течение болезни бывает очень тяжёлым, и к лечению нужно подходить ответственно, поскольку на кону не только качество жизни, но и она сама: по статистике 90–95 % пациентов не дотягивают до 40–50 лет
В общем, единственными обследованиями были ЭКГ, ЭХО, холтеровское мониторирование (круглосуточная ЭКГ в условиях обычной повседневной активности, когда датчики ЭКГ приклеивают к телу. — Прим. ред.), посещения кардиолога, поддерживающая терапия и совет «не болеть». Следовать ему у меня не очень-то получалось. В тринадцать лет, пролежав дома две недели с температурой под сорок, я всё же попала в инфекционную больницу — и в приёмном отделении врач, увидев моё горло, побледнела и объявила моей маме, что у меня дифтерия. Мама расплакалась, а меня перевели в отделение и поместили в палату с выздоравливающими от гриппа — «чудесное» решение. Хорошо, что диагноз не подтвердился и дифтерии у меня всё же не было. Тем не менее любая болезнь, тем более поражающая дыхательные пути, негативно отражается на сердце, что в моём случае просто опасно.
Всю жизнь я прожила с диагнозом «дисплазия соединительной ткани», а синдром Марфана был лишь фенотипом — это значит, что у меня были проявления синдрома, но он не был подтверждён генетически. По мнению некоторых врачей, особенно тех, кто видел меня впервые, он отсутствовал вовсе — ведь полного набора классических симптомов не было. У меня всё более или менее нормально с костями и состоянием позвоночника, на месте хрусталик глаза; болезнь можно заподозрить только из-за патологий сердечно-сосудистой системы, высокого роста, арахнодактилии (те самые «паучьи пальцы») и повышенной эластичности. Я и сейчас, рассказывая врачам об анамнезе, упоминаю дисплазию чаще, чем синдром Марфана — иначе они просто вежливо кивают и пропускают мимо ушей.
Поскольку меня не беспокоила боль, я жила как обычный подросток. За моё сердце переживала мама, но неполная осведомлённость помогла пройти этот путь гораздо легче. Если бы тогда она была в курсе всех сюрпризов, которые может преподнести болезнь, не знаю, как справлялась бы с этим. Я даже рада, что и сейчас она знает не больше прежнего — теперь уже я думаю о её сердце. Реально же подтвердить диагноз удалось только на тридцатом году жизни: я самостоятельно сдала анализ на мутации генов в рамках так называемой панели заболеваний соединительной ткани. С его помощью выявили мутацию, характерную для синдрома Марфана, однако не в «горячих точках» — возможно, поэтому я и не типичный представитель болезни.
Синдром Марфана — это генетическое заболевание, но оно не обязано проявляться у кого-то из родни. У моих родителей, например, нет никаких проявлений, ни внешне, ни «внутренне». В основе заболевания лежат мутации в гене, отвечающем за синтез фибриллина — важнейшего структурного белка межклеточного матрикса, придающего эластичность и сократимость соединительной ткани. При нём сильнее всего страдают органы с наибольшей «концентрацией» соединительной ткани: сердце, глаза, спина, связки. Синдром не лечится, и любая терапия направлена на конкретные пострадавшие органы — например, я должна пить курсами таблетки, помогающие сердечно-сосудистой системе. Течение болезни бывает очень тяжёлым, и к лечению нужно подходить ответственно, поскольку на кону не только качество жизни, но и она сама: по статистике 90–95 % пациентов не дотягивают до 40–50 лет.
Ограничения в первую очередь касаются физических нагрузок. Нельзя заниматься профессиональным спортом, хотя по иронии судьбы данных у меня как раз предостаточно — например, высокий рост и колоссальная гибкость (до сих пор могу закинуть ногу за голову или сесть в позу лотоса с разбега). Людям с синдромом Марфана показан разумный спорт без резких движений, например плавание. В путешествиях моя аптечка не больше, чем у обычного человека, никакой специфики — но тут дело скорее не в факте синдрома, а в степени его проявления.
Из-за «кукольного театра», как назвали ширму между мной и оперирующими врачами, я слышала, как акушер постоянно повторяла ассистентам: «Не рвите ткани, не рвите ткани!»
Со всей серьёзностью проблемы я столкнулась, когда мы с мужем захотели ребёнка. Генетик приговорил к суррогатному материнству или возможной беременности после кардиохирургии, к которой не было прямых показаний. Мне объясняли, что функции сердца достаточно для моего собственного жизнеобеспечения, но при беременности нагрузка удваивается. Я же настаивала, что при относительно лёгкой степени поражения сердца и сосудов нормальное течение беременности возможно. Я побывала во всех кардиоцентрах Москвы, пролила море слёз и всё же получила разрешение, при условии постоянного наблюдения кардиолога. Так, через два месяца я пришла вставать на учёт в кардиологический перинатальный центр, который согласился меня вести.
Конечно, решиться на беременность было очень страшно, особенно после неоднозначного прогноза врачей — моя аорта находится пока на докритическом уровне. Надеюсь, там она и останется — до критического лишь несколько миллиметров. Надежду вселила врач-гинеколог, посвятившая полжизни изучению дисплазии соединительной ткани. После разговора с ней я поняла, что если я не попробую выносить ребёнка вопреки своему желанию, то буду жалеть всю жизнь. Где-то глубоко внутри я взяла на себя этот риск и не жалею.
Беременность прошла отлично, а дочь родилась через плановое кесарево сечение немного раньше срока. Врачи безумно боялись, что аорта «рванёт» от максимальных нагрузок и я умру прямо в роддоме, где была самой «тяжёлой» роженицей. Дочь — моя победа, и мы назвали её Викторией. Из-за «кукольного театра», как назвали ширму между мной и оперирующими врачами, я слышала, как акушер постоянно повторяла ассистентам: «Не рвите ткани, не рвите ткани!» — но в тот момент я была готова ко всему, лишь бы с ребёнком всё было в порядке. Операция длилась в два раза дольше обычного, врач была мокрой, будто на неё вылили ведро воды. Я пару раз почти теряла сознание, в чувства приводил анестезиолог, а потом, уже в реанимации, я узнала, что потеряла почти литр крови. Состояние моей сердечно-сосудистой системы осталось без изменений, то есть таким же, как до беременности.
Оглядываясь на свою юность, я понимаю, что мне в какой-то степени повезло, если можно так выразиться. Да, меня не обошли высокий рост, пластинка на зубах, одно плечо выше другого — конечно, я выделялась из толпы, были насмешки среди сверстников и слёзы ночами в ванной комнате. Но такое было у многих, такова подростковая жизнь. После общения с мамами детей с более тяжёлым синдромом Марфана, я поняла, что моя жизнь могла бы быть гораздо труднее.
Свой оптимизм я взрастила сама, по крупинкам — при этом я заядлый параноик, что вообще присуще людям с синдромом Марфана. В интернете есть множество статей и информации, в которой очень легко запутаться и накрутить себя ещё сильнее. Специалистов же, которые разбираются в проблеме, единицы. Есть группы и форумы, где люди описывают свои симптомы, делятся опытом и даже «ставят» себе и другим диагнозы; многие хорошо изучили проблему и дадут в этой области фору некоторым докторам. Но большинство пишут о неизбежности, о моральной и физической боли, о доживании — поэтому я в этих группах не сижу, не хочу загонять себя в переживания ещё глубже. Конечно, я понимаю свои перспективы, но всегда ищу положительное в окружающем мире, стараюсь не зацикливаться на проблемах — иначе крайне сложно выбраться из разрастающейся паники. Конечно, не нужно закрывать глаза на свой диагноз, будто его нет — он есть, и очень опасен, но это не клеймо и не приговор.
О моей особенности знают лишь самые близкие, и многие люди из окружения задают вопросы о втором ребёнке. Но я не могу решиться на этот шаг, просто не имею права. Ещё до рождения дочери я взяла на себя колоссальную ответственность за неё и перед ней. Возможно, мне придётся столкнуться с кардиохирургией, и мне очень страшно. Мой организм с годами даёт о себе знать всё больше, количество визитов к врачам ежегодно растёт — но это не повод сидеть и считать оставшиеся дни. Мне бывает очень сложно. Мысли о судьбе дочери, которой уже два года, и о продолжительности собственной жизни порой неделями не дают спать, но я делаю всё возможное, чтобы минимизировать проявления синдрома — ведь если я сдамся, лучше не станет. Нужно жить свою жизнь, а не проживать её.
Фотографии: spacedrone808 — stock.adobe.com (1, 2, 3)
Источник
Приветствую, дорогие читатели. Сегодня хотел бы поговорить про высоких людей с Синдромом Марфана, для которых высокий рост – это не просто особенность организма, а симптом серьезной болезни, требующей к себе постоянного внимания.
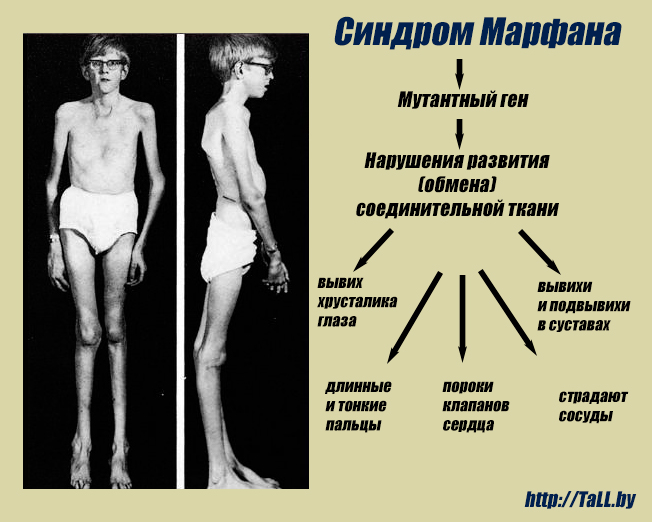
Синдром Марфана — заболевание наследственного типа, при котором поражается соединительная ткань с вовлечением в процесс скелетно-мышечной системы и глаз. Установлено, что причиной патологии является мутация гена фибриллина FBN1. Заболевание полиморфно — может протекать с разной выраженностью клинической картины, и характеризуется появлением все новых типов мутации в генах.
Синдром Марфана получил своё название от фамилии французского педиатра А. Марфана, который вперые представил описание 5-летней девочки Габриель с необычными, непрерывно прогрессирующими аномалиями скелета, и дал патологии своё имя.
Распространенность синдрома — 1 случай на 10000 человек. Риск рождения ребенка с синдромом Марфана повышается после достижения отцом возраста 35 лет и достигает 50% при наличии патологии у одного из родителей. Врожденная аномалия наследуется по аутосомно-доминантному типу. В ее основе лежит дефект важнейшего гена, отвечающего за синтез коллагена.

Во время внутриутробного развития происходит нарушение формирования волокон соединительной ткани, утеря ими прочности, в результате чего волокна не способны выдерживать естественные нагрузки. Поэтому наибольшие атипичные изменения претерпевают крупные сосуды, клапаны сердца, связки глаза, твердое небо, скелет и мышцы.
Без адекватной терапии продолжительность жизни людей с синдромом Марфана не более 40 лет. Терапия позволяет увеличить этот срок вдвое и более.
История заболевания
В 1876 г. симптомы неизвестной патологии были отмечены доктором Вильямсом, но клинические наблюдения проводились гораздо позже — в 1896 г. педиатром из Франции А. Марфаном. Врач в течение 5-ти лет оценивал состояние девочки с неизученными ранее аномалиями, заключающимися в прогрессировании дистрофии скелета и мышечной ткани.
К середине 20-го века имелось множество описанных случаев, когда у больных наблюдались симптомы, близкие к патологии Марфана, и все они относились к заболеваниям наследственного типа. Среди таких случаев — расслоение аорты, пороки сердца, эктопия хрусталиков, сопровождающиеся деформацией костей (грудной клетки, позвоночника) и внешними отклонениями от нормы (высокий рост, худоба, длинные конечности). Американским генетиком МакКьюсиком было проведено детальное исследование мутаций хромосом и открыта новая группа заболеваний соединительной ткани.

Симптомы синдрома Марфана
Многообразие вариантов генетической мутации обуславливает различные формы течения болезни. Нередко они малозаметны, иногда приводят к инвалидизации человека в раннем возрасте. Частый признак синдрома Марфана — высокий рост (до 200 см.), при этом туловище непропорционально короткое, а конечности удлиненные и тонкие. Пальцы у больных длинные, паукообразные (арахнодактилия) Из-за недоразвития подкожной клетчатки и мышечной дистрофии страдающие синдромом Марфана имеют астеническое телосложение.
Прочие внешние симптомы патологии (в каждом индивидуальном случае может наблюдаться один или несколько из них):
— гиперподвижность суставов;
— аномалии строения тазобедренного сустава;
— кифоз, сколиоз;
— вывихи шейного сегмента позвоночника;
— деформация грудной клетки;
— плоскостопие;
— глубокая посадка глаз;
— уменьшенная нижняя челюсть, нарушение роста зубов;
— высокое нёбо;
— атрофические «растяжки» на коже;
— паховые грыжи, частые разрывы связок.
Более серьезные изменения при синдроме Марфана протекают в организме. Самые тяжелые из них развиваются со стороны сердца и сосудов и могут привести к смерти ребенка еще на первом году жизни. Среди них:
— дефекты ветвей легочной артерии, аорты (расширения, аневризмы, расслоения);
— пороки сердца (чаще — поражения клапанов);
— стенозы артерий.
Подобные нарушения вызывают тахикардию, мерцательную аритмию вплоть до фибрилляции предсердий или развития сердечной недостаточности.
Со стороны глаз наблюдается выраженная миопия, вывих хрусталика, аномалии развития роговицы, уменьшение в размерах радужки, косоглазие, патологии сосудистой стенки сетчатки. При прогрессирующем вывихе хрусталика или при отслойке сетчатки уже в раннем возрасте больные могут полностью потерять зрение.
Со стороны нервной системы при синдроме Марфана происходит растяжение твердой мозговой оболочки и выбухание ликвора в костные дефекты в пояснично-крестцовом отделе позвоночника (дуральная эктазия). Легкие страдают гораздо реже, так как незначительные нарушения их работы не оказывают влияния на дыхательную функцию. Но в отдельных случаях снижение эластичности альвеол может привести к спонтанному пневмотораксу, развитию дыхательной недостаточности. Прочими симптомами патологии могут быть эктопия почек, деформации мочевого пузыря, половых органов.
Лечение и профилактика осложнений
Специфической терапии заболевания не существует: изменить гены еще до рождения ребенка невозможно. Лечение только симптоматическое и зависит от тех изменений в организме, которые развиваются у больного синдромом Марфана. Некоторые осложнения патологии можно успешно корректировать, другие — устранять оперативным путем.

Пациент должен наблюдаться у группы специалистов — офтальмолога, невролога, кардиолога, ортопеда, хирурга. Основное направление терапии — поддержка функций сердца и сосудов.
Методы лечения:
— прием препаратов (адреноблокаторы, антиаритмические лекарства, антикоагулянты и т.д.);
— хирургия пороков сердца (дисфункции клапанов, расширения, расслоения легочной артерии), аорты, протезирование клапанного аппарата.
Нормализация зрения проводится при помощи коррекции миопии (ношение очков, линз), лечения катаракты, глаукомы, имплантации искусственного хрусталика.
При поражении суставов и позвоночника проводится оперативное лечение (протезирование, пластика суставов, устранение межпозвоночных грыж), выправление кифоза, сколиоза при помощи тракции, мануальной терапии. Из медикаментозных средств используются миорелаксанты, витамины группы В. Также применяется физиолечение, занятия ЛФК.
При поражении легких часто требуется хирургическое вмешательство (дренирование их полости).
Беременность больными синдромом Марфана должна строго планироваться и развиваться под контролем группы врачей, специализирующихся на лечении людей с подобными патологиями. Родоразрешение — только при помощи кесарева сечения. Еще до наступления беременности желательно обследоваться на предмет возможного прогрессирования расслойки аорты и, по возможности, провести операцию по замене части сосуда. Консультация генетика позволит рассчитать примерный риск по передаче заболевания по наследству.
Известные люди с синдромом Марфана
Хоть синдром Марфана – очень редкое заболевание, есть немало знаменитостей, больных синдромом Марфан: Фло Хайман (призер Олимпийских игр по волейболу), Джон Тавенер (композитор), Джоуи Рамон (музыкант), Лесли Хорнби (фотомодель и певица) и другие.
Среди исторических личностей, известных во всем мире, с синдромом Марфана можно выделить:

Музыкант-скрипач Никколо Паганини. Поскольку Паганини умер еще до описания синдрома, данные о его заболевании исследуются по сохранившимся изображениям и дневнику лечащего врача. Скрипач имел характерную деформацию пальцев, высокий рост и худобу, непропорциональное развитие конечностей, впалую грудь, мышечную слабость.
Писатель Ганс Кристиан Андерсен. Имел угловатое лицо, был очень худым и длинноруким, рано заполучил проблемы со зрением.
Президент Америки Авраам Линкольн. Кроме всех внешних признаков синдрома Марфана у Линкольна наблюдались ревматические боли, «разболтанность» суставов, но, в то же время — хорошая физическая выносливость.
Писатель Корней Чуковский. Наличие большого непропорционального носа, длинных конечностей не помешало Чуковскому стать одним из лучших творцов современности и доктором филологии.
Осама Бен Ладен. «Террорист №1» мира имел высокий рост и малый вес и большие проблемы с суставами и позвоночником, а также вытянутый череп и слишком узкое лицо.
Источники и ссылки:
1. Причины высокого роста — статья, в которой рассмотрены основные причины высокого роста человека.
2. Синдром Марфана в википедии — о синдроме Марфана в свободной энциклопедии.
3. Русскоговорящее сообщество людей с синдромом Марфана — форум о синдроме Марфана, общение, обсуждение болезни.
4. Международный фонд синдрома Марфана — фонд помощи больным с синдромом Марфана.
5. Группа вконтакте для больных синдромом Марфана.
Источник