Синдром марфана этиология и патогенез

Синдром Марфана: причины, диагностика, лечениеЭтиология и встречаемость синдрома Марфана. Синдром Марфана (MIM №154700) — панэтническое аутосомно-доминантное заболевание соединительной ткани, вызванное мутациями в гене фибриллина 1 (FBN1, MIM № 134797). Синдром Марфана имеет встречаемость около 1 на 10 000. Приблизительно 25-35% пациентов имеют новую мутацию. Мутации, вызывающие синдром Марфана, разбросаны по гену, и каждая мутация обычно уникальна в семье. Патогенез синдрома МарфанаГен FBN1 кодирует фибриллин 1, внеклеточный матричный гликопротеид с широким распределением. Фибриллин 1 полимеризуется, формируя микрофибриллы как в эластичных, так и в неэластичных тканях, например стенке аорты, цилиарных поясках и коже. Мутации влияют на синтез, процессинг, секрецию, полимеризацию или устойчивость фибриллина 1. Исследования накопления и экспрессии фибриллина 1 в культуре клеток предположили доминантный отрицательный патогенез, т.е. синтез мутантного фибриллина 1 тормозит образование нормальных микрофибрилл нормальным фибриллином 1 или стимулирует протеолиз несоответствующих внеклеточных микрофибрилл. Последние исследования на мышиных моделях синдрома Марфана указывают, что половинного количества нормального фибриллина 1 недостаточно, чтобы проводить эффективную сборку микрофибрилл. Таким образом, патогенезу болезни также может содействовать гаплонедостаточность. Кроме синдрома Марфана, мутации в гене FBN1 могут вызывать другие синдромы, включая неонатальный синдром Марфана, изолированные скелетные симптомы, аутосомно-доминантную эктопию хрусталиков и фенотип MASS (марфаноидные симптомы, включая пролапс митрального клапана или миопию, пограничное и непрогресирующее расширение аорты, и неспецифические изменения скелета и кожи). В общем, фенотипы довольно схожи в пределах семьи, хотя тяжесть фенотипических проявлений может значительно изменяться. До настоящего времени точное соотношение между генотипом и фенотипом не определено. Внутрисемейная и межсемейная изменчивость позволяет предполагать, что в определении фенотипа важную роль играют окружающая среда и эпигенетические факторы. Последние исследования на мышиных моделях показывают, что фибриллин 1 не просто структурный белок, и что синдром Марфана не просто результат структурной слабости тканей. Более того, микрофибриллы фибриллина 1 в норме связывают и уменьшают концентрацию и активность факторов роста суперсемейства TGFb. Потеря фибриллина 1 увеличивает сигналы свободного TGFb значительно содействующие заболеванию, так как антагонисты TGFb устраняют легочные и клапанные изменения, наблюдаемые у мышей с недостаточностью фибриллина 1.
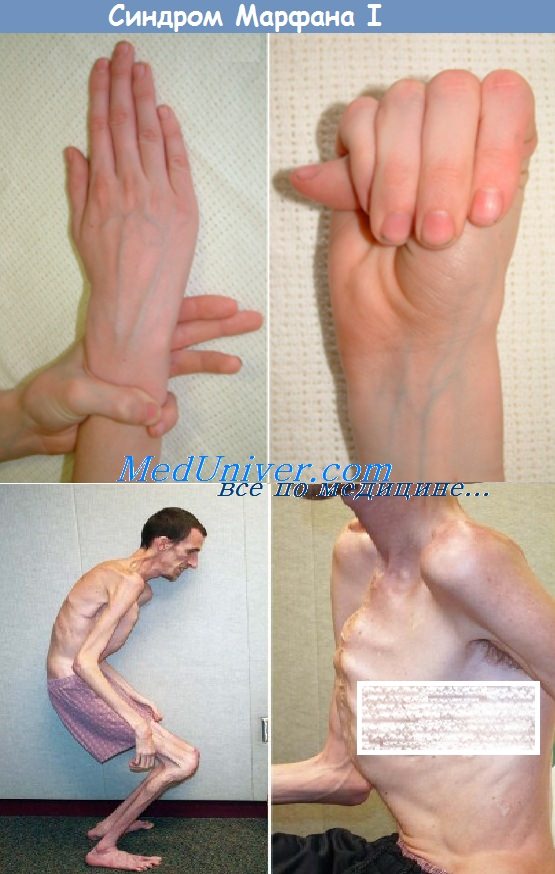
Фенотип и развитие синдрома МарфанаСиндром Марфана — мультисистемное заболевание со скелетными, глазными, сердечно-сосудистыми, легочными, кожными и другими аномалиями. Скелетные аномалии включают очень высокий рост (отношение размаха рук к росту >1,05; соотношение верхнего и нижнего сегментов <0,85 у взрослых), арахнодактилию, аномалии грудины, сколиоз, разболтанность суставов, готическое нёбо. Аномалии глаз включают подвывих хрусталиков, уплощение роговицы, удлинение глазного яблока и гипоплазию радужки. Сердечнососудистые аномалии включают пролапс митрального клапана, аортальную регургитацию и расширение и расслаивающую аневризму восходящей аорты. Легочные аномалии включают спонтанный пневмоторакс и расширение концевых пузырьков. Аномалии кожи включают атрофические бороздки и рецидивирующие грыжи. Аномалии твердой мозговой оболочки включают выбухание оболочки в крестцово-поясничном отделе. Большинство признаков синдрома Марфана появляются с возрастом. Скелетные аномалии типа аномалии грудины и сколиоза ухудшаются с ростом костей. Подвывих хрусталика часто присутствует уже в раннем детстве, но может развиваться и в юности. С повышенной частотой при синдроме Марфана встречаются отслойка сетчатки, глаукома и катаракты. Сердечно-сосудистые осложнения обнаруживаются в любом возрасте и развиваются в течение всей жизни. Основные причины преждевременной смерти пациентов с синдромом Марфана — сердечная недостаточность вследствие регургитации клапанов и аневризмы и разрыва аорты. Тем не менее в связи с улучшением хирургической и терапевтической помощи при аневризме аорты выживание улучшилось. С 1972 по 1993 г. ожидаемый возраст выживания для 50% пациентов поднялся с 49 до 74 лет для женщин и с 41 до 70 лет для мужчин. Особенности фенотипических проявлений синдрома Марфана:
Лечение синдрома МарфанаСиндром Марфана — клинический диагноз, определяемый по наличию конкретных симптомов. Подтверждение синдрома Марфана идентификацией мутаций в гене FBN1 в настоящее время практически нецелесообразно, поскольку крайняя аллельная гетерогенность делает идентификацию причинно-обусловленной мутации в каждой семье крайне трудозатратной, а также из-за недостаточно надежной корреляции между генотипом и фенотипом. Анализ мутаций имеет недостаточную чувствительность или специфичность для синдрома Марфана, что ограничивает его клиническую пользу. Для синдрома Марфана недоступно эффективное лечение; следовательно, помощь сфокусирована на профилактике осложнений и симптоматическом лечении. Оказание офтальмологической помощи включает регулярные осмотры, коррекцию миопии и, часто, замену хрусталика. Ортопедическая помощь заключается в укрепляющем лечении или хирургической коррекции сколиоза. Помощь при аномалии грудины в основном косметическая. Физиотерапия может компенсировать нестабильность суставов. Сердечно-сосудистые проблемы решаются комбинацией терапевтических и хирургических мероприятий. Терапевтические усилия направлены на предохранение или замедление развития расширения корня аорты за счет уменьшения кардиологических показателей, снижения артериального давления и усилия выброса желудочков с помощью бета-адреноблокаторов, ограничение участия в контактных видах спорта, соревновательных видах спорта и в изометрических упражнениях. Профилактическая замена корня аорты показана, когда расширение аорты или аортальная регургитация становится достаточно тяжелой. Большинству пациентов в настоящее время проводят надклапанную замену корня аорты, не требующую постоянного приема противосвертывающих препаратов. Гемодинамические изменения, связанные с беременностью, могут приводить к прогрессирующему расширению или расслоению аорты. Полагают, что расслоение аорты вторично к гормональным изменениям, увеличению объема крови и сердечного выброса, связанных с беременностью и родами. Современные исследования считают, что риск беременности слишком велик, если ширина корня аорты превышает 4 см. Женщины могут выбрать проведение надклапанной замены аорты перед беременностью. Риски наследования синдрома МарфанаПациенты с синдромом Марфана имеют 50% риск иметь ребенка, больного синдромом Марфана. В семьях, передающих синдром Марфана, членов семьи, находящихся в группе риска, можно выявлять, либо обнаруживая мутацию (в тех редких случаях, когда она известна), либо анализом сцепления, если маркеры, тесно сцепленные с локусом FBN1, имеют очевидную связь с болезнью в семье пробанда. Пренатальная диагностика доступна только для семей, в которых возможны исследования сцепления или известна мутация в гене FBN1. Пример синдрома Марфана. Д.Л., здоровый 16-летний ученик средней школы, звезда баскетбола, направлен в генетическую клинику для обследования по поводу синдрома Марфана. Телосложение Д.Л. похоже на телосложение его отца. Отец Д.Л., высокий субтильный человек, умер во время утренней пробежки; у других членов семьи случаев скелетных аномалий, внезапной смерти, снижения зрения или врожденных аномалий не было. При медицинском осмотре выявлены астеническое телосложение, высокое дугообразное нёбо, небольшая деформация грудины по типу «куриной» груди, арахнодактилия, соотношение размах рук/рост 1,1, диастолический шум и стрии на плечах и бедрах. Эхокардиография выявила расширение корня аорты с аортальной регургитацией. Офтальмологическое обследование показало двусторонний иридодонез и легкое смещение хрусталиков кверху. На основе медицинского осмотра и результатов обследования генетик объяснил пациенту и его матери, что у него синдром Марфана. — Также рекомендуем «Синдром Миллера-Дикера: причины, диагностика, лечение» Оглавление темы «Наследственные болезни»:
|
Источник
Врожденные аномалии, особенно те, которые передаются по аутосомно-доминантному типу, впечатляют своим разнообразием. Одним из таких недугов является синдром Марфана. Для данной болезни характерно наличие целого комплекса выраженных клинических симптомов. Лечение недуга специфическое, а потому требует детального рассмотрения.
Источник: https://vseolady.ru/sindrom-marfana.html
Что это за недуг
Синдром Марфана – что это такое? Это генетическое заболевание, возникающее на фоне мутации гена, отвечающего за синтез фибриллина. Этот белок является важной структурной единицей соединительной ткани. Именно благодаря ему, она обладает известной способностью к растяжению и сокращению, без появления разрывов.
Нехватка фибриллина – это прямой путь к патологии соединительной ткани. Волокна формируются неправильно, они не такие прочные, эластичные и упругие. Вследствие этого, ткань неспособна выдерживать традиционные для человека нагрузки. Она может быстро и в большом количестве разрываться, на месте повреждений появляются характерные рубцы – стрии. Растяжки на спине у подростков, в паху, под руками и прочих частях тела могут появляться именно из-за проблем с развитием соединительной ткани.
В 75% диагностированных случаях болезнь имела семейный тип наследования, и только в 25 процентах наблюдалась первичная мутация. Есть гипотеза, что недуг передается ребенку преимущественно от отца. Если у него или у его родственников был поставлен такой диагноз, вероятность того, что болезнь передастся ребенку, существенно повышается.
Синдром Марфана – генетическое заболевание, возникающее на фоне мутации гена, отвечающего за синтез фибриллинаОтличительной особенностью этого заболевания является то, что оно может поражать сразу несколько систем человеческого организма. Люди с синдромом Марфана выглядят соответствующим образом – у них, как правило, высокий рост, но маленькое туловище, из-за чего конечности (как нижние, так и верхние) выглядят непропорционально длинными. Пальцы тоже очень длинные, из-за чего их нередко называют паучьими, наблюдается вдавленная грудная клетка. Из-за натянутой кожи кажется, что больные намного моложе, чем есть на самом деле.
Синдром Марфана – что это за болезнь:
Форма заболеванияТяжесть протеканияХарактер течения
Выделяют две основных формы синдрома:
- Стертая – клинические проявления незначительные, и наблюдаются преимущественно в одной, максимум – в двух системах организма (например, сердечно-сосудистой и опорно-двигательного аппарата).
- Выраженная – симптоматика хорошо выражена в одной, двух, трех или больше системах. Также к такой форме относят тип недуга, когда симптоматика выражена слабо, но она затрагивает больше двух систем человеческого организма.
Данное заболевание может протекать в легкой, средней и тяжелой форме. Чем тяжелее течение, тем сильнее выражены симптомы, и тем больше поражение организма. В самых крайних случаях функциональность организма нарушена практически полностью, пациенту грозит летальный исход.Синдром может быть стабильным, то есть, таковым, при котором клинические проявления остаются примерно на одном уровне – не утихают, но и не усиливаются. При прогрессирующей форме негативные признаки заболевания постоянно усиливаются, постепенно ухудшая состояние больного.
Синдром Марфана – симптомы
Поскольку аутоимунный недуг затрагивают сразу несколько систем человеческого организма, нет ничего странного в том, что признаков очень много. Некоторые из них выражены лучше, другие – хуже. Ряд симптомов схож с проявлениями других заболеваний, например:
- гомоцистинурия;
- врожденная контрактурная арахнодактилия (синдром Билса);
- наследственная артроофтальмопатия (синдром Стиклера);
- MASS-синдром;
- синдромы Элерса-Данлоса, Лойса-Дитца, Шпринцена-Голдберга;
- семейная эктопия хрусталика.
Проявляющую симптоматику можно разделить на несколько групп, что поможет лучше понять ее особенности.
- Опорно-двигательный аппарат
Как уже говорилось выше, больные этим заболеванием имеют преимущественно высокий рост, короткое туловище и длинные конечности с такими же пальцами. Отмечается долихоцефалия – длинный и узкий лицевой скелет. Мышцы и подкожная клетчатка развиты крайне слабо. У некоторых больных присутствует нарушение прикуса и высокое, аркообразное небо.
Источник: https://vseolady.ru/sindrom-marfana.html
Помимо этого, признаки синдрома Марфана со стороны скелетной системы также включают в себя:
- килевидная или воронкообразная грудная клетка;
- плоскостопие;
- кифосколиотическая деформация позвоночника;
- нарушения функции суставов (гипермобильность);
- протрузия вертлужной впадины.
Симптомы нарушений функциональности опорно-двигательного аппарата являются одними из основных показателей для постановки правильного диагноза, поскольку они, как правило, хорошо выражены, и присутствуют у подавляющего количества пациентов.
- Мышечная система
К классическим проявлениям данного заболевания в данном случае можно отнести мышечную гипотонию, гипоплазию жировой ткани, практически полное отсутствие мускулатуры, а также небольшую массу тела. Это плохо для всех людей, но для беременных женщин – особенно, так как их кожа практически не растягивается, и под ней, по сути, ничего нет. Проблема в том, что подобные изменения характерны и для многих других недугов. Соответственно, здесь нужно либо дифференцировать диагноз, либо обращать внимание на другие симптомы.
- Сердечно-сосудистая система
Например, те, которые проявляются со стороны сердечно-сосудистой системы. Как правило, они доминируют, а изменения, затронувшие эту систему, чаще всего определяют исход заболевания. Преимущественно страдает сердце, однако патологии актуальны и для сосудов. Симптомов здесь может быть очень много, и практически все они несут определенную угрозу здоровью и даже жизни пациента:
- вегетативные расстройства;
- незначительные признаки гипертрофии миокарда левого желудочка и предсердия;
- проблемы с внутрижелудочковой проходимостью;
- поражение митрального клапана;
- патологическое удлинение и разрыв створочных хорд;
- нарушения ритма, тахикардия, фибрилляция;
- инфекционный эндокардит;
- прогрессирующее расширение восходящей части и клапанного кольца аорты;
- аневризмы.
На фоне проблем с сердцем и сосудами наблюдается и мышечная слабость. Человек чувствует себя постоянно разбитым, минимальные физические нагрузки даются ему с большим трудом. Данное заболевание может также привести к образованию врожденных пороков сердца – стеноза легочной артерии, коарктации аорты и т.д.
- Зрительная система
У многих больных наблюдаются проблемы и с органами зрения. В первую очередь, к традиционным симптомам можно отнести:
- миопия (более известна, как близорукость);
- эктопия (подвывих хрусталика);
- уплощение и увеличение размера роговицы;
- гипоплазия радужной оболочки – аниридия;
- расширение или сужение сосудов глаза;
- косоглазие;
- афакия;
- колобома.
Основная проблема в том, что негативные проявления со стороны зрительной системы наблюдаются уже с самого раннего возраста и, как правило, постоянно прогрессируют. Если не придавать этому факту должного значения, то это приведет к существенному ухудшению функции зрения, вплоть до слепоты.
- Нервная система
В первую очередь, это эктазия твердой мозговой оболочки, в т. ч. пояснично-крестцовое менингоцеле, анизокория, пирамидные расстройства, асимметрия сухожильных рефлексов. Могут быть и другие проявления – это еще до сих пор изучается современной наукой.
Признаки синдромаДля данного заболевания характерны и другие проблемы с внутренними органами и тканями, например:
- спонтанный пневмоторакс;
- эмфизема легких;
- дыхательная недостаточность;
- атрофические стрии;
- паховые и бедренные грыжи, которые постоянно рецидивируют;
- вывихи и разрывы связок;
- эктопия почек;
- опущение мочевого пузыря и матки;
- варикозное расширение вен.
Диагностика синдрома Марфана и лечение
Диагностировать это заболевание можно с помощью семейного анамнеза, наблюдения за клиническими симптомами, физического осмотра пациента, а также на основе данных, собранных посредством соответствующих диагностических и лабораторных мероприятий:
- электрокардиограмма;
- ЭхоКГ;
- анализы крови;
- офтальмологический осмотр;
- генетические исследования;
- рентген;
- функциональная диагностика;
- компьютерная томография;
- магнитно-резонансная терапия;
- аортография.
Поскольку причины симптома Марфана изучены достаточно хорошо, главная задача врачей – это дифференцировать данное заболевание от других патологий. Это очень важно, поскольку ряд симптомов актуален не только для этого синдрома, но и многих других болезней. Только после постановки окончательного диагноза можно приступать к терапевтическим мероприятиям.
Лечение синдрома Марфана направлено, в первую очередь, на исправление и профилактику патологий со стороны систем человеческого организма. Особенное внимание уделяют сердечно-сосудистой системе, так как ее поражение влечет за собой серьезную угрозу жизни пациента. лечение может быть консервативным или хирургическим. Решение о выборе оптимального способа принимает лечащий врач, ориентируясь на текущее состояние больного.
Нужно понимать, что этот недуг является генетическим, то есть, его нельзя вылечить.
https://youtu.be/5O5F21XWzWo
Пациенту нужно следить за тем, чтобы свести к минимуму риск поражения тех или иных органов, а также устранить уже существующие проблемы. Благодаря этому, существенно повысится качество его жизни.
Хирургическое лечение проводится преимущественно на сердце и сосудах, а также на глазах. При правильном подходе к борьбе с этим синдромом, продолжительность жизни пациента можно увеличить до 70 лет, что является очень хорошим показателем.
Источник: https://vseolady.ru/sindrom-marfana.html
Источник