Синдром марфана фото больных людей на ранней стадии

Что это за болезнь?
Синдром Марфана – это наследственное заболевание, передающееся по аутосомно-доминантному типу и характеризуется поражением соединительной ткани и ее компонентов.
Болезнь Марфана вызывается мутированием гена, кодирующего фибриллин -1.
Люди с синдромом Марфана имеют удлиненные конечности, паукообразные пальцы и слабый (недоразвитый) подкожно-жировой слой и сверхгибкие суставы (см. фото ниже).
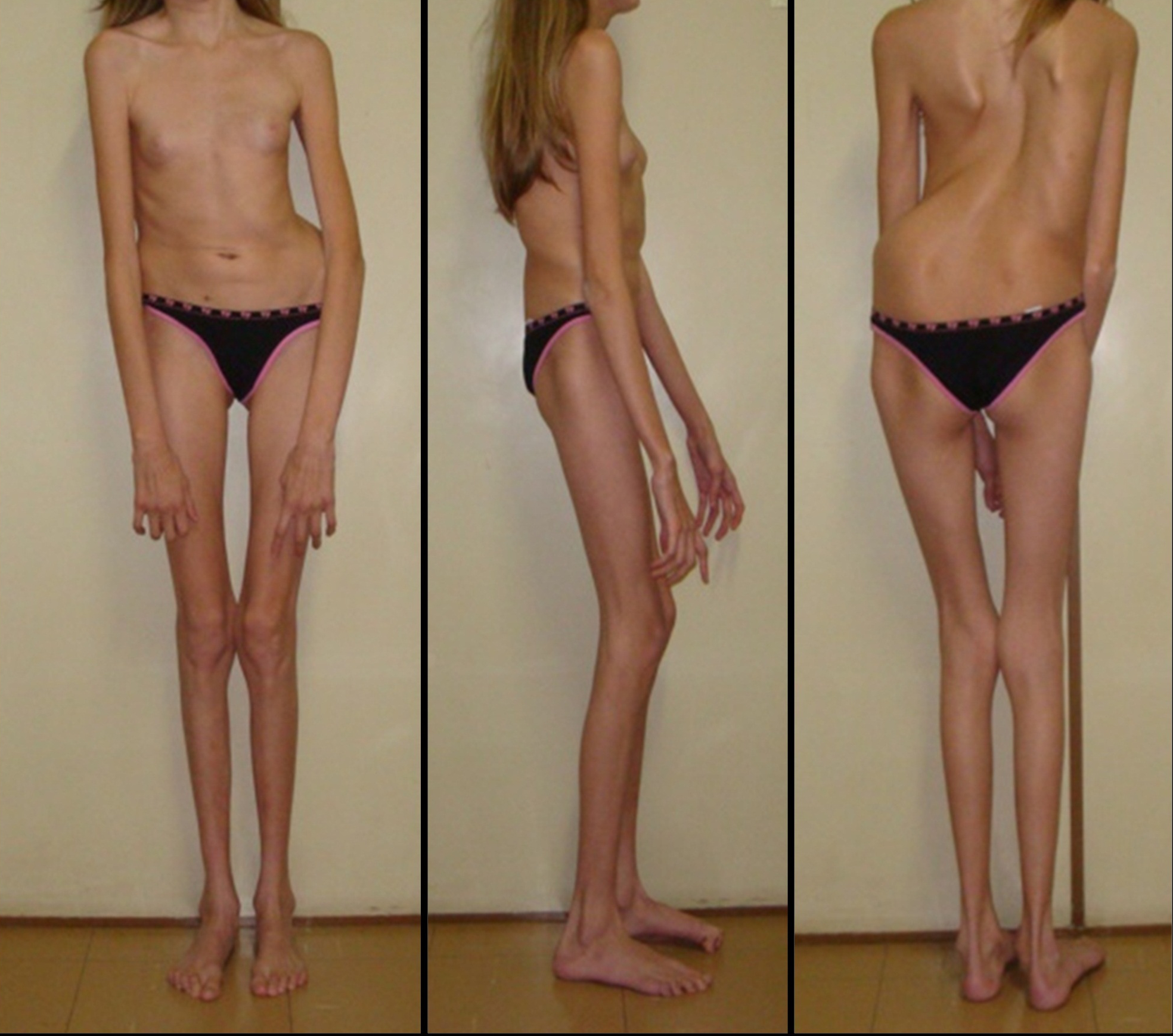
Кроме изменений костно-суставной системы, характерны изменения зрительного анализатора и сердечно-сосудистой системы. Также возможно поражение нервной, дыхательной и других систем.
Впервые описал данную патологию Вильямс, который заметил у своих брата и сестры – выпадение хрусталика, при этом они были очень высокими и имели гипермобильные суставы. Затем заметил Марфан, врач – невролог, у которого в течение 20 лет наблюдалась женщина с подобными симптомами, а затем еще 20 детей.
Причины возникновения

Болезнь Марфана у детей наследуется по аутосомно-доминантному типу (т. е передается от родителя к ребенку).
Также возможны мутации за счет воздействия на организм женщины факторов внешней среды (ионизирующее излучение, лучевая терапия, радиация).
Причины возникновения и механизм развития заболевания недостаточно изучены.
Особая роль отводится нарушению процессов обмена, в результате которых накапливается в коллагеновых и эластических волокнах большое количество мукополисахаридов.
Это ведет к тому, что соединительная ткань перерастягивается, легко подвергается механическому воздействию и приводит к развитию клинической симптоматики.
Классификация
Выделяют следующие формы болезни Марфана:
В зависимости от генной предрасположенности:
- семейная (патология передается от родителя к ребенку);
- спорадическая (патология вызвана внезапным мутированием в геноме).
В зависимости от проявлений клиники:
- стертая, когда признаки заболевания практически не проявляются и могут быть не замечены в течение всей жизни. Патологические изменения выявляются в одной или двух системах.
- выраженная, когда признаки заболевания касаются двух и более органов и систем (сердце, кости и суставы, легкие, кожа, глаза).
Симптомы болезни Марфана
Синдромом Марфана у людей ведет к их выделению в обществе своим непропорциональным строением скелета. Для новорожденных на ранней стадии болезни, характерны длинные пальцы на руках, а к 7-9 годам у детей формируется развернутая клиническая картина.
У взрослых характерна различная симптоматика в зависимости от системы поражения:
- Нервная система: болезненность в поясничной области, головные боли,поражение симпатической и парасимпатической иннервации органов брюшной полости и малого таза (слабость кишечной стенки, недержание мочевого пузыря у ребенка). Также высок риск развития инсульта, субарахноидального кровоизлияния и разрыва аневризм головного мозга.
- Сердечно – сосудистая система: пороки сердца (сужение легочной артерии, пролабирование створок двустворчатого клапана, дилатационная кардиомиопатия, расширение границ сердца (аорты и всех ее отделов), дефекты МЖП и МЖЖ перегородок. У больных может развиваться нарушение ритма и проводимости в виде аритмий.
- Опорно-двигательный аппарат: телосложение астенической формы (дети худые), высокий рост у мужчин 190±10 см, у женщин 179±8 см, слаборазвитый подкожно-жировой слой, длинные пальцы (паукообразные), плоскостопие, череп и лицо вытянутые и узкие, недоразвитость скул, нарушение развития зубов и прикуса, вытянутая нижняя челюсть, готическое верхнее небо, гипермобильность суставов (смотрите на картинки выше). С возрастом ребенка может прогрессировать деформация позвоночного столба, с развитием сколиоза. Также может деформироваться грудная клетка, образуется вдавление – «грудь сапожника». Деформированный тазобедренный сустав нередко приводит к инвалидности при неоказанном своевременном лечении.
- Орган зрения: смещение хрусталика за счет слабого связочного аппарата) на ранней стадии, уплощение роговицы, развитие близорукости или дальнозоркости, спазм аккомодации, отслоение сетчатки.
- Кожа и мягкие ткани: перерастяжение кожи с образованием стрий атрофического характера. Они возникают внезапно, не связаны с колебание веса людей, беременностью и гормональным фоном. Кожа липкая, потная, с мраморным оттенком. Подкожно-жировой слой слабо развит, поэтому у больных наблюдаются грыжевые выпячивания в области передней брюшной стенки.
- Дыхательная система: развитие буллезной эмфиземы легких, проявляющейся кашлем, одышкой, развитием дыхательной недостаточности и спонтанного пневмоторакса.
Другие признаки:
- развитие опущения почек (нефроптоз);
- выпадение органов малого таза (опущение матки у женщин, или ее полное выпадение);
- варикоз вен нижних конечностей;
- запоры.
Диагностика
Диагностика основана на тщательном сборе анамнеза заболевания, выраженности клинической картины, данных осмотра, на результатах лабораторных и инструментальных методов исследований.
Сбор анамнеза включает в себя: наличие в семье данной патологии (родители, братья, сестры) или наличие факторов, провоцирующих мутацию в геноме человека.
К лабораторным методам относят: анализ генотипа ДНК с мутирующим геном, определение гликозаминогликанов в моче.
К инструментальным методам исследования относят:
- ЭКГ, служит для обнаружения патологии сосудов и сердца (ССС). Выявляют характерные нарушения ритма и проводимости в виде мерцательной аритмии, желудочковой экстрасистолии, развитие дилатационной гипертрофии миокарда левого желудочка.
- ЭхоКГ, также служит для обнаружения патологии ССС. Выявляют расширение аорты и ее структур, пролабирование двустворчатого клапана, увеличение размеров левой половины сердца.
- УЗИ сердца проводится для определения осложнений (расслаивающаяся аневризма).
- Рентген органов грудной клетки (изменения скелета, расширение полостей сердца, корней легких и др.)
- Компьютерная томография, магнитно-резонансно-ядерная томография позволяет выявить патологии костно-суставной, нервной системы, нарушение кровообращения в сосудах головного и спинного мозга.
Данные методы исследования служат для обнаружения критериев синдрома Марфана в различных органах и системах. Они играют самую важную роль для постановки и подтверждения диагноза, а в последующем, для определения тактики лечения.
Существуют следующие критерии диагностики синдрома Марфана:
| Система | Большие критерии | Малые критерии |
| Опорно- двигательный аппарат Должны быть: 4 больших критерия, либо 2 больших и 1 малый. |
|
|
| Орган зрения | Смещение хрусталика | Уплощенная роговица, близорукость, дальнозоркость, недоразвитие радужки и цилиарной мышцы глаз. |
| Сердечно-сосудистая система | Расширение аорты и ее структур | Пролабирование двустворчатого клапана, расширение клапана легочной артерии у лиц, не достигших 40 лет, отложение солей кальция на створках двустворчатого клапана, расслаивание аорты. |
| Дыхательная система | Отсутствуют | Внезапно развивающийся пневмоторакс (скопление воздуха в грудной клетке), верхушечные буллы. |
| Кожа | Отсутствуют | Повторное развитие грыжевых выпячиваний, атрофические стрии. |
| Нервная система | Расширения сосудов оболочек спинного мозга в поясничномкрестцовом отделе позвоночного столба. | Отсутствуют |
| Генетические изменения | Наличие данных критериев у родителей, детей, братьев, сестер, бабушек, дедушек. Наличие мутирующего гена, кодирующего фибриллин 1. | Отсутствуют |
Для постановки диагноза «Синдром Марфана» учитывается один признак из перечня больших критериев или малый критерий, характерный каждой из пораженной систем, кроме опорно-двигательного аппарата, (необходимо, как минимум, 4 критерия), а также наличие в семейном анамнезе больных с данной патологией.
Лечение синдрома Марфана
От синдрома Марфана полностью избавиться и устранить механизм его развития невозможно. Лечение базируется на улучшении общего состояния больного, устранении клинических проявлений и проведении профилактических мероприятий, препятствующих развитию осложнений.
Больным с данным синдромам рекомендовано ограничить физическую нагрузку до низкого уровня, или минимального. Риск появления патологии сердечно-сосудистой системы возрастает при средних и высоких физических нагрузках.
Следует обходить стороной и повседневные нагрузки, при которых возможно повышение внутригрудного давления, ведущему к развитию пневмоторакса (например, подъем тяжестей, подъем по этажам).
Больные с синдромом Марфана должны консультироваться у разных специалистов, в зависимости от клинически пораженных систем органов. Следует проходить медицинские осмотры каждые полгода в течение всей жизни.
Медикаментозное лечение
Медикаментозная терапия направлена на устранение клинической картины заболевания.
Со стороны сердечно-сосудистой системы рекомендованы β-адреноблокаторы (например: Анаприлин), которые снижают скорость распространения пульсовых волн при стремительно растущем расширении аорты и обратного тока крови на двустворчатом клапане или клапане аорты.
β-адреноблокаторы также оказывают положительный эффект при нарушении ритма и проводимости, в сочетании с сердечными гликозидами.
Но следует помнить о существующих противопоказаниях данных групп препаратов:
- хронический обструктивный бронхит;
- бронхиальная астма;
- снижение частоты сердечных сокращений;
- низкое артериальное давление.
Блокаторы каналов кальция используются при наличии противопоказаний к В-адреноблокаторам.
Хирургия
Хирургическое лечение проводится, если есть осложнения со стороны сердечно-сосудистой системы, с целью коррекции пораженных участков. Его проводят при пролабировании двустворчатого клапана и расслаивании аорты.
При этом осуществляется протезирование двустворчатого клапана.
У беременных с тяжелым течением болезни Марфана, роды разрешаются хирургическим путем.
Профилактика
С профилактической целью, во избежание развития инфекционных осложнений, образования тромбов и тромбоэмболии, назначаются антикоагулянты (гепарин), антибактериальная терапия и витаминотерапия.
- При синдроме Марфана с тяжелым поражением зрительного анализатора проводится хирургическая коррекция зрения, после которой пациенты должны носить очки или контактные линзы.
- Если возникают осложнения, проводят лазерную коррекцию глаукомы, катаракты, удаляют смещаемый хрусталик, заменяя его на искусственный.
- При функциональной дисфункции опорно-двигательного аппарата возникает необходимость стабилизирования позвоночника с помощью металлических пластин.
- При выраженной деформировании грудной клетки проводится торакопластика.
- При протрузии тазобедренных суставов производят внутреннее протезирование суставов.
Прогноз
Длительность жизни в среднем при синдроме Марфана составляет 30-45 лет.
Известно, что это многие знаменитые личности страдали данным синдромом. Это и Ганс Христиан Андерсен – датский писатель, автор знаменитой Русалочки; Авраам Линкольн – 16 президент США, Майкл Феллпс- известный пловец, многократный олимпийский чемпион. А также известные композиторы – Никколо Паганини, Сергей Рахманинов.
Люди с данной патологией должны тщательно следить за своим здоровьем, постоянно наблюдаться и консультироваться со своим лечащим врачом, избегать чрезмерных физических нагрузок.
По мимо медикаментозного лечения, необходимо проведение профилактических мероприятий с целью улучшения общего самочувствия, повышения иммунитета, соответствующий режим труда и отдыха.
Видеозаписи по теме
Источник
В последние годы отмечается значительный рост наследственных заболеваний с поражением соединительной ткани. Некоторые специалисты считают, что причина такого явления кроется в накоплении в процессе эволюции человека новых генетических изменений (мутаций), к чему приводят внутренние и внешние факторы окружающей среды. Другие полагают, что на самом деле просто возросли диагностические возможности в связи с совершенствованием медико-генетических знаний, которые позволяют распознать генетическую патологию и поставить правильный диагноз.
Большую группу наследственных заболеваний составляет поражение соединительной ткани. Так как она является неотъемлемой частью всех органов и систем в организме, то такие патологии отличаются множеством нарушений и клинических симптомов. Одним из самых известных генетических заболеваний с поражением соединительной ткани считается синдром Марфана.
Такая патология характеризируется большой вариабельностью симптоматики (от скрытых форм до несовместимых с жизнью вариантов течения) и чаще всего включает поражения сердечно-сосудистой системы, глаз, центральной нервной системы и опорно-двигательного аппарата.
Встречается заболевание достаточно редко – 1 случай на 10 000 человек. Но если обратиться к статистике европейских стран, то частота значительно выше – 1-3 случая на 5000 человек, что связано с большей доступностью специфической диагностики скрытых форм патологии.

Известные люди тоже болели синдромом Марфана
Причины и генетика патологии
Впервые болезнь детально описал в 1896 году французский педиатр А.Марфан, в честь которого и назвали патологию. Он наблюдал за 5-летней девочкой астенического телосложения с непропорционально длинными конечностями и врожденной арахнодактилией (длинными пальцами). К середине 20-х годов прошлого столетия уже имелось множество подобных описанных клинических случаев у детей и взрослых. Американский генетик Мак Кьюсик провел детальное исследование мутаций хромосом и открыл новую группу наследственных заболеваний соединительной ткани, куда и отнесли синдром Марфана.
По этиологии синдром Марфана является генетическим заболеванием с аутосомно-доминантным типом наследования, с различной степенью экспрессивности (клинических проявлений генетических изменений). Примерно 85% случаев недуга носит наследственный характер, то есть наследуется от родителей. Остальные случаи являются новыми, то есть возникают вследствие новых спонтанных мутаций, а не передаются по наследству.
Непосредственная причина патологии в 95% случаев – это мутация в гене, которые кодирует строение фибриллина-1 и/или фибриллина-2. Локализируется мутация гена FBN1 и FBN2 в хромосоме 15 и 3.

Синдром Марфана наследуется по аутосомно-доминантному типу
Фибриллин – это основа эластических волокон соединительной ткани гликопротеиновой природы. Он составляет каркас межклеточного вещества, сосудистых стенок, хрящей, хрусталика глаза и многих других органов и тканей. В случае наличия описанной мутации у пациента соединительная ткань отличается повышенной способностью к растяжению, становится менее прочной и выносливой к механическим воздействиям, что и становится причиной клинических проявлений синдрома.
Примерно в 5% случаев непосредственной причиной синдрома Марфана (атипичные формы патологии) является точковая мутация гена, который кодирует строение α2-цепи коллагена первого типа.
Классификация
Согласно МКБ-10, синдром Марфана входит в класс врожденных дефектов развития и хромосомных патологий, тут патологию можно найти под шифром Q87.4.
Детальной клинической классификации недуга на сегодняшний день не существует, но выделяют несколько форм болезни, в зависимости от тех или иных критериев.
В зависимости от выраженности симптомов:
- стертая форма – признаки патологии мало выражены и могут оставаться незамеченными на протяжении всей жизни, как правило, изменения касаются не более 2 систем органов;
- клинически выраженная форма – симптомы патологии хорошо заметны и встречаются более чем в 2 системах органов.

Внешний вид ребенка, больного синдромом Марфана
В зависимости от генетического фактора:
- семейная форма диагностируется в случаях, когда болезнь передается по наследству;
- спорадическая форма определяется тогда, когда патология обусловлена новой спонтанной мутацией у индивида и при этом не встречается у его родственников.
Симптомы синдрома Марфана
Признаки синдрома Марфана очень разнообразны. Поэтому их рассматривают с точки зрения поражения отдельных органов и систем:
- опорно-двигательного аппарата;
- органов зрения;
- сердечно-сосудистой системы;
- нервной системы;
- дыхательной системы;
- кожных покровов и мягких тканей;
- прочих органов и систем.
Опорно-двигательный аппарат
Патологическая соединительная ткань обусловливает развитие ряда специфических фенотипических признаков и деформаций скелета у пациентов с данной патологией.

Характерный внешний вид пациентов с синдромом Марфана
Как правило, внешне пациенты с синдромом Марфана выглядят достаточно специфически. Для них характерно:
- астеническое телосложение;
- высокий рост;
- плохое развитие подкожной жировой клетчатки, из-за чего люди выглядят худощавыми;
- очень длинные верхние и нижние конечности при относительно коротком туловище;
- череп вытянутый (долихоцефалический);
- удлиненные пальцы – паукообразные (арахнодактилия);
- лицо узкое, вытянутое по вертикали;
- готическое верхнее небо;
- недоразвитие скул;
- выступающая нижняя челюсть (прогнатизм);
- неправильный рост (скученность) зубов и патологический прикус;
- гипермобильность суставов, их «разболтанность;
- глубоко посажены в черепе глаза.

Пример того, как определить наличие арахнодактилии, характерной для синдрома Марфана
По мере роста ребенка могут появляться различные деформации скелета. Чаще всего появляются искривления позвоночного столба. У пациентов диагностируют сколиоз, патологический кифоз и лордоз, осанку по типу «прямой спины» (сглаживание физиологического поясничного лордоза). Также появляются подвывихи и вывихи в шейном отделе позвоночника, примерно у 20% больных диагностируют поясничный спондилолистез.

Выраженная степень арахнодактилии при синдроме Марфана со специфическими контрактурами пальцев
Среди других скелетных деформаций встречается:
- протрузия вертлужной впадины тазобедренного сустава 2-3 степени, которая становится причиной диспластического коксартроза и инвалидности многих пациентов, если не выполнить операцию эндопротезирования;
- килевидная и вдавленная деформация грудной клетки;
- плоскостопие (продольное и поперечное).
Для пациентов в СМ также характерна остеопения (снижение минеральной плотности костей) и частые патологические переломы костей на ее фоне, а также склонность к привычным вывихам, например, плеча.

Вдавленная деформация грудной клетки у пациента с синдромом Марфана и результат ее хирургической коррекции
Сердечно-сосудистая система
Среди поражений кардиоваскулярной системы при СМ чаще всего встречаются:
- пролапс створок митрального клапана с регургитацией или без
- миксаматоз сердца;
- дилятационная кардиомиопатия с развитием сердечной недостаточности;
- аневризмы аорты и других сосудов (мозговых, почечных, пр.);
- расширение легочной артерии и различных отделов аорты.
Именно кардиоваскулярные патологические изменения при СМ определяют прогноз и продолжительность жизни пациентов. Примерно 90% всех пациентов с данной генетической патологией умирают в возрасте 40-50 лет вследствие таких осложнений, как расслоение и разрыв аневризмы аорты, других сосудов, прогрессирующей недостаточности сердца вследствие дилатации его камер и изменений клапанного аппарата.
При наличии СМ возможно наличие врожденных пороков сердца у детей. Чаще всего встречаются коарктация аорты, стеноз (сужение) легочной артерии, дефект межжелудочковой и межпредсердной перегородки.
Также такие пациенты склонны к различным сердечным аритмиям, среди которых и опасные для жизни (мерцательная аритмия, желудочковая тахикардия и экстрасистолия), к инфекционному эндокардиту.

Аневризмы аорты и их разрыв чаще всего становятся причиной смерти пациента с синдромом Марфана
Орган зрения
Патологические изменения глаз являются весьма характерными для данного недуга. Примерно у 60-80% пациентов диагностируется дислокация хрусталика из-за слабости его связочного аппарата, причем еще в младенческом возрасте. Среди других характерных признаков:
- уплощение роговицы;
- увеличение размеров глазного яблока в длину;
- миопия или гиперметропия;
- нарушение процесса аккомодации из-за недоразвития цилиарной мышцы.
В случае выявления описанных поражений органа зрения у новорожденного ребенка следует задуматься о возможной генетической патологии.

Дислокация хрусталика – типичный признак синдрома Марфана
Нервная система
Из-за патологического строения стенок сосудов у пациентов с СМ повышен риск геморрагических инсультов, а также кровоизлияний в мозг при разрыве сосудистых аневризм, субарахноидальных кровотечений.
Среди аномалий развития встречается эктазия твердой мозговой оболочки. Чаще всего приходится сталкиваться с пояснично-крестцовой эктазией мозговой оболочки (выпячивание твердой мозговой оболочки за пределы позвоночного канала через дефект в строении позвонков). Это большой критерий СМ, который встречается в 40% случаев заболевания.
У части пациентов встречаются отклонения в интеллектуальном развитии, но большинство людей с СМ характеризируются высокими показателями IQ.
Органы системы дыхания
В большинстве случаев изменения бронхолегочного аппарата диагностируются случайно. Характерно развитие булл в верхних частях легких, которые иногда могут разрываться с развитием спонтанного пневмоторакса.
Также из-за деформаций грудной клетки пациенты склонны к развитию эмфиземы легких, частых инфекционных заболеваний органов дыхания и дыхательной недостаточности.

Буллезная эмфизема может быть причиной спонтанного пневмоторакса у пациентов с синдромом Марфана
Кожа и мягкие ткани
Встречается повышенная растяжимость кожного покрова, которая сочетается с развитием атрофических стрий. Последние появляются спонтанно, они никак не связаны с колебанием веса, беременностью или гормональными нарушениями. Подкожный жир выражен слабо у пациентов с синдромом Марфана. Они часто страдают рецидивирующими грыжами передней брюшной стенки.
Встречаются и многие другие патологические симптомы поражения прочих органов и тканей при СМ. Например, опущение почек (нефроптоз), выпадение мочевого пузыря и матки у женщин, варикозное расширение вен, хронические запоры и др.

Диагностика синдрома Марфана
Диагностика при синдроме Марфана носит в основном клинический характер. Обязательно учитывают анамнез, в том числе и семейный (наличие подобных проблем у кого-то из родственников), данные объективного обследования и осмотра. Также проводят множество дополнительных диагностических процедур для выявления патологии тех или иных органов и систем. Для этого применяют ЭКГ, УЗИ сердца и сосудов, рентгенографию органов грудной клетки, КТ, МРТ внутренних органов, позвоночника, головного мозга, офтальмоскопию и прочие исследования органа зрения, аортографию, ангиографию и много других методик, в зависимости от клинической ситуации и симптомов болезни.
Существуют общепринятые диагностические критерии синдрома Марфана (большие и малые), которые позволяют с большой степенью вероятности поставить правильный, но предварительный диагноз пациенту.

Окончательный диагноз синдрома Марфана выставляют только после анализа генотипа (ДНК-диагностика) и выявления специфической мутации в гене, ответственном за продукцию фибриллина, с помощью молекулярно-генетических методик.
Лечение заболевания
К сожалению, на сегодняшний день вылечить синдром Марфана, как и повлиять на его причину, невозможно. Терапия в основном направлена на улучшение качества жизни больного человека, устранение симптомов и профилактику осложнений.
Лечение синдрома Марфана должно быть комплексным и может включать как консервативные, так и хирургические методики.
Всем пациентам с СМ рекомендуют ограничения в физической активности к среднему или низкому уровню, избегать тяжелого физического труда, не заниматься спортом, так как это способствует прогрессированию патологии и травматизму.
Больные должны наблюдаться у различных докторов: кардиолога, окулиста, травматолога-ортопеда, клинического генетика, невролога и др.

Лечением синдрома Марфана должна заниматься целая команда специалистов
В случае выявления тех или иных кардиоваскулярных проблем назначают комплексное лечение, медикаменты для профилактики осложнений. Проводят медикаментозную коррекцию аритмий, частоты сердечных сокращений, артериального давления. Обязательно всем пациентам назначают прием бета-блокаторов, если нет противопоказаний к этой группе средств, которые имеют протекторный эффект в отношении возможного расслоения аорты.
В случае патологии клапанного аппарата сердца, аневризме аорты и других сосудов, расслоении их стенок может понадобиться операция. Также хирургическое вмешательство используют для коррекции деформаций опорно-двигательного аппарата.
Рекомендуемые специалистами схемы терапии больных с СМ в обязательном порядке включают коллаген-образующие и метаболические средства, препараты для восполнения макро- и микроэлементов.
В случае проблем с органом зрения также проводят оперативные вмешательства при эктопии хрусталика, лазерную коррекцию зрения, подбирают очки или контактные линзы.
В целом подбор лечебных методик и спектр применяемых средств очень индивидуальны. Это полностью зависит от присутствующих симптомов и степени выраженности болезни у конкретного пациента.

Основная задача в лечении пациентов с Синдромом Марфана заключается в предотвращении сердечно-сосудистых повреждений
Прогноз
Синдром Марфана отличается, как правило, хроническим прогрессирующим течением. Продолжительность жизни пациентов при условии полноценного комплекса лечебных мероприятий в среднем составляет 45 лет. Основными факторами риска преждевременной смерти выступают осложнения, которые возникают вследствие патологии кардиоваскулярной системы.
Синдром Марфана и беременность
Пациентки с СМ могут иметь детей, причем здоровых, но это очень опасно по двум причинам:
- Беременная женщина имеет очень высокий риск летальных осложнений СМ со стороны сердечно-сосудистой системы. Так как при вынашивании ребенка создается усиленная нагрузка на организм матери, а особенно на сердце и сосуды, то очень большие шансы у пациенток с синдромом Марфана получить разрыв аневризмы, ее формирование, расслоение аорты и прочие смертельно опасные осложнения.
- Риск передачи данной наследственной патологии своему ребенку составляет 50%.
Поэтому специалисты не рекомендуют беременеть женщинам с синдромом Марфана.
Возможна ли профилактика болезни?
К сожалению, специфической профилактики синдрома Марфана не существует. Семейная пара, в которой один или оба родителя страдают данной патологией, в обязательном порядке должны планировать беременность и пройти медико-генетическое консультирование у врача-генетика. Вместе с этим можно проводить пренатальную (дородовую) диагностику на предмет наличия СМ у плода.
Источник