Синдром марфана что это за болезнь фото

Что это за болезнь?
Синдром Марфана – это наследственное заболевание, передающееся по аутосомно-доминантному типу и характеризуется поражением соединительной ткани и ее компонентов.
Болезнь Марфана вызывается мутированием гена, кодирующего фибриллин -1.
Люди с синдромом Марфана имеют удлиненные конечности, паукообразные пальцы и слабый (недоразвитый) подкожно-жировой слой и сверхгибкие суставы (см. фото ниже).
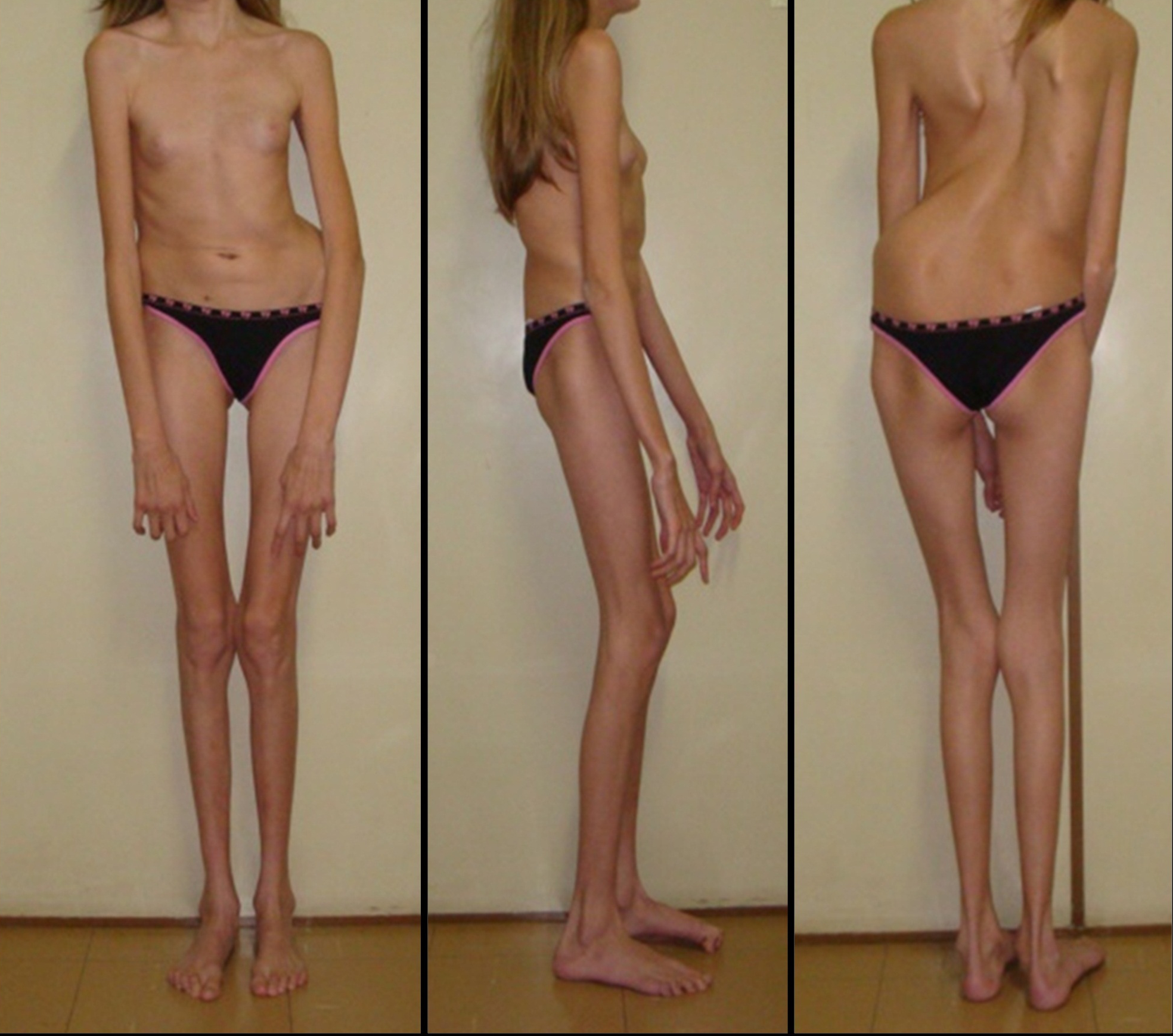
Кроме изменений костно-суставной системы, характерны изменения зрительного анализатора и сердечно-сосудистой системы. Также возможно поражение нервной, дыхательной и других систем.
Впервые описал данную патологию Вильямс, который заметил у своих брата и сестры – выпадение хрусталика, при этом они были очень высокими и имели гипермобильные суставы. Затем заметил Марфан, врач – невролог, у которого в течение 20 лет наблюдалась женщина с подобными симптомами, а затем еще 20 детей.
Причины возникновения

Болезнь Марфана у детей наследуется по аутосомно-доминантному типу (т. е передается от родителя к ребенку).
Также возможны мутации за счет воздействия на организм женщины факторов внешней среды (ионизирующее излучение, лучевая терапия, радиация).
Причины возникновения и механизм развития заболевания недостаточно изучены.
Особая роль отводится нарушению процессов обмена, в результате которых накапливается в коллагеновых и эластических волокнах большое количество мукополисахаридов.
Это ведет к тому, что соединительная ткань перерастягивается, легко подвергается механическому воздействию и приводит к развитию клинической симптоматики.
Классификация
Выделяют следующие формы болезни Марфана:
В зависимости от генной предрасположенности:
- семейная (патология передается от родителя к ребенку);
- спорадическая (патология вызвана внезапным мутированием в геноме).
В зависимости от проявлений клиники:
- стертая, когда признаки заболевания практически не проявляются и могут быть не замечены в течение всей жизни. Патологические изменения выявляются в одной или двух системах.
- выраженная, когда признаки заболевания касаются двух и более органов и систем (сердце, кости и суставы, легкие, кожа, глаза).
Симптомы болезни Марфана
Синдромом Марфана у людей ведет к их выделению в обществе своим непропорциональным строением скелета. Для новорожденных на ранней стадии болезни, характерны длинные пальцы на руках, а к 7-9 годам у детей формируется развернутая клиническая картина.
У взрослых характерна различная симптоматика в зависимости от системы поражения:
- Нервная система: болезненность в поясничной области, головные боли,поражение симпатической и парасимпатической иннервации органов брюшной полости и малого таза (слабость кишечной стенки, недержание мочевого пузыря у ребенка). Также высок риск развития инсульта, субарахноидального кровоизлияния и разрыва аневризм головного мозга.
- Сердечно – сосудистая система: пороки сердца (сужение легочной артерии, пролабирование створок двустворчатого клапана, дилатационная кардиомиопатия, расширение границ сердца (аорты и всех ее отделов), дефекты МЖП и МЖЖ перегородок. У больных может развиваться нарушение ритма и проводимости в виде аритмий.
- Опорно-двигательный аппарат: телосложение астенической формы (дети худые), высокий рост у мужчин 190±10 см, у женщин 179±8 см, слаборазвитый подкожно-жировой слой, длинные пальцы (паукообразные), плоскостопие, череп и лицо вытянутые и узкие, недоразвитость скул, нарушение развития зубов и прикуса, вытянутая нижняя челюсть, готическое верхнее небо, гипермобильность суставов (смотрите на картинки выше). С возрастом ребенка может прогрессировать деформация позвоночного столба, с развитием сколиоза. Также может деформироваться грудная клетка, образуется вдавление – «грудь сапожника». Деформированный тазобедренный сустав нередко приводит к инвалидности при неоказанном своевременном лечении.
- Орган зрения: смещение хрусталика за счет слабого связочного аппарата) на ранней стадии, уплощение роговицы, развитие близорукости или дальнозоркости, спазм аккомодации, отслоение сетчатки.
- Кожа и мягкие ткани: перерастяжение кожи с образованием стрий атрофического характера. Они возникают внезапно, не связаны с колебание веса людей, беременностью и гормональным фоном. Кожа липкая, потная, с мраморным оттенком. Подкожно-жировой слой слабо развит, поэтому у больных наблюдаются грыжевые выпячивания в области передней брюшной стенки.
- Дыхательная система: развитие буллезной эмфиземы легких, проявляющейся кашлем, одышкой, развитием дыхательной недостаточности и спонтанного пневмоторакса.
Другие признаки:
- развитие опущения почек (нефроптоз);
- выпадение органов малого таза (опущение матки у женщин, или ее полное выпадение);
- варикоз вен нижних конечностей;
- запоры.
Диагностика
Диагностика основана на тщательном сборе анамнеза заболевания, выраженности клинической картины, данных осмотра, на результатах лабораторных и инструментальных методов исследований.
Сбор анамнеза включает в себя: наличие в семье данной патологии (родители, братья, сестры) или наличие факторов, провоцирующих мутацию в геноме человека.
К лабораторным методам относят: анализ генотипа ДНК с мутирующим геном, определение гликозаминогликанов в моче.
К инструментальным методам исследования относят:
- ЭКГ, служит для обнаружения патологии сосудов и сердца (ССС). Выявляют характерные нарушения ритма и проводимости в виде мерцательной аритмии, желудочковой экстрасистолии, развитие дилатационной гипертрофии миокарда левого желудочка.
- ЭхоКГ, также служит для обнаружения патологии ССС. Выявляют расширение аорты и ее структур, пролабирование двустворчатого клапана, увеличение размеров левой половины сердца.
- УЗИ сердца проводится для определения осложнений (расслаивающаяся аневризма).
- Рентген органов грудной клетки (изменения скелета, расширение полостей сердца, корней легких и др.)
- Компьютерная томография, магнитно-резонансно-ядерная томография позволяет выявить патологии костно-суставной, нервной системы, нарушение кровообращения в сосудах головного и спинного мозга.
Данные методы исследования служат для обнаружения критериев синдрома Марфана в различных органах и системах. Они играют самую важную роль для постановки и подтверждения диагноза, а в последующем, для определения тактики лечения.
Существуют следующие критерии диагностики синдрома Марфана:
| Система | Большие критерии | Малые критерии |
| Опорно- двигательный аппарат Должны быть: 4 больших критерия, либо 2 больших и 1 малый. |
|
|
| Орган зрения | Смещение хрусталика | Уплощенная роговица, близорукость, дальнозоркость, недоразвитие радужки и цилиарной мышцы глаз. |
| Сердечно-сосудистая система | Расширение аорты и ее структур | Пролабирование двустворчатого клапана, расширение клапана легочной артерии у лиц, не достигших 40 лет, отложение солей кальция на створках двустворчатого клапана, расслаивание аорты. |
| Дыхательная система | Отсутствуют | Внезапно развивающийся пневмоторакс (скопление воздуха в грудной клетке), верхушечные буллы. |
| Кожа | Отсутствуют | Повторное развитие грыжевых выпячиваний, атрофические стрии. |
| Нервная система | Расширения сосудов оболочек спинного мозга в поясничномкрестцовом отделе позвоночного столба. | Отсутствуют |
| Генетические изменения | Наличие данных критериев у родителей, детей, братьев, сестер, бабушек, дедушек. Наличие мутирующего гена, кодирующего фибриллин 1. | Отсутствуют |
Для постановки диагноза «Синдром Марфана» учитывается один признак из перечня больших критериев или малый критерий, характерный каждой из пораженной систем, кроме опорно-двигательного аппарата, (необходимо, как минимум, 4 критерия), а также наличие в семейном анамнезе больных с данной патологией.
Лечение синдрома Марфана
От синдрома Марфана полностью избавиться и устранить механизм его развития невозможно. Лечение базируется на улучшении общего состояния больного, устранении клинических проявлений и проведении профилактических мероприятий, препятствующих развитию осложнений.
Больным с данным синдромам рекомендовано ограничить физическую нагрузку до низкого уровня, или минимального. Риск появления патологии сердечно-сосудистой системы возрастает при средних и высоких физических нагрузках.
Следует обходить стороной и повседневные нагрузки, при которых возможно повышение внутригрудного давления, ведущему к развитию пневмоторакса (например, подъем тяжестей, подъем по этажам).
Больные с синдромом Марфана должны консультироваться у разных специалистов, в зависимости от клинически пораженных систем органов. Следует проходить медицинские осмотры каждые полгода в течение всей жизни.
Медикаментозное лечение
Медикаментозная терапия направлена на устранение клинической картины заболевания.
Со стороны сердечно-сосудистой системы рекомендованы β-адреноблокаторы (например: Анаприлин), которые снижают скорость распространения пульсовых волн при стремительно растущем расширении аорты и обратного тока крови на двустворчатом клапане или клапане аорты.
β-адреноблокаторы также оказывают положительный эффект при нарушении ритма и проводимости, в сочетании с сердечными гликозидами.
Но следует помнить о существующих противопоказаниях данных групп препаратов:
- хронический обструктивный бронхит;
- бронхиальная астма;
- снижение частоты сердечных сокращений;
- низкое артериальное давление.
Блокаторы каналов кальция используются при наличии противопоказаний к В-адреноблокаторам.
Хирургия
Хирургическое лечение проводится, если есть осложнения со стороны сердечно-сосудистой системы, с целью коррекции пораженных участков. Его проводят при пролабировании двустворчатого клапана и расслаивании аорты.
При этом осуществляется протезирование двустворчатого клапана.
У беременных с тяжелым течением болезни Марфана, роды разрешаются хирургическим путем.
Профилактика
С профилактической целью, во избежание развития инфекционных осложнений, образования тромбов и тромбоэмболии, назначаются антикоагулянты (гепарин), антибактериальная терапия и витаминотерапия.
- При синдроме Марфана с тяжелым поражением зрительного анализатора проводится хирургическая коррекция зрения, после которой пациенты должны носить очки или контактные линзы.
- Если возникают осложнения, проводят лазерную коррекцию глаукомы, катаракты, удаляют смещаемый хрусталик, заменяя его на искусственный.
- При функциональной дисфункции опорно-двигательного аппарата возникает необходимость стабилизирования позвоночника с помощью металлических пластин.
- При выраженной деформировании грудной клетки проводится торакопластика.
- При протрузии тазобедренных суставов производят внутреннее протезирование суставов.
Прогноз
Длительность жизни в среднем при синдроме Марфана составляет 30-45 лет.
Известно, что это многие знаменитые личности страдали данным синдромом. Это и Ганс Христиан Андерсен – датский писатель, автор знаменитой Русалочки; Авраам Линкольн – 16 президент США, Майкл Феллпс- известный пловец, многократный олимпийский чемпион. А также известные композиторы – Никколо Паганини, Сергей Рахманинов.
Люди с данной патологией должны тщательно следить за своим здоровьем, постоянно наблюдаться и консультироваться со своим лечащим врачом, избегать чрезмерных физических нагрузок.
По мимо медикаментозного лечения, необходимо проведение профилактических мероприятий с целью улучшения общего самочувствия, повышения иммунитета, соответствующий режим труда и отдыха.
Видеозаписи по теме
Источник
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 6 марта 2016;
проверки требует 71 правка.
Синдром (болезнь) Марфана — аутосомно-доминантное заболевание из группы наследственных патологий соединительной ткани. Синдром вызван мутацией гена, кодирующего синтез гликопротеина фибриллина-1, и является плейотропным. Заболевание характеризуется различной пенетрантностью и экспрессивностью. В классических случаях лица с синдромом Марфана высоки (долихостеномелия), имеют удлинённые конечности, вытянутые пальцы (арахнодактилия) и недоразвитие жировой клетчатки. Помимо характерных изменений в органах опорно-двигательного аппарата (удлинённые трубчатые кости скелета, гипермобильность суставов), наблюдается расширение аорты и/или эктопия хрусталика.
Диагностика синдрома Марфана (СМ) сегодня основана на Гентских критериях (DeРаере A. et al.,1996) и пересмотра Гентских критериев в 2010 году. В основу алгоритма диагностики положено выделение больших и малых критериев, характеризующих выраженность изменений соединительной ткани в различных органах и системах.
Большие критерии свидетельствуют о наличии в соответствующей системе патологически значимых изменений. Малые критерии (а в некоторых случаях – один большой критерий) свидетельствуют о вовлечении той или иной системы в патологию соединительной ткани
Без лечения продолжительность жизни лиц с синдромом Марфана часто ограничивается 30—40 годами[2], и смерть наступает вследствие разрыва аневризмы аорты или застойной сердечной недостаточности. В странах с развитым здравоохранением больные успешно лечатся и доживают до преклонного возраста.
Наследственное заболевание.
Эпидемиология[править | править код]
Синдром Марфана — редкое заболевание с классическим менделевским наследованием. Распространённость в популяции составляет порядка 1 на 5000. Синдром диагностируется во всем мире, в любых этнических группах. Мужчины и женщины страдают с одинаковой частотой[3].
История[править | править код]
Впервые признаки заболевания были описаны в 1875 году американским офтальмологом Э. Вильямсом (англ. E. Williams), описавшим эктопию хрусталика у брата и сестры, которые были исключительно высокими и имели гипермобильные суставы от рождения[4]. В последующие годы эта болезнь наблюдалась французским профессором педиатрии Антуаном Марфаном, который представил в 1896 году клиническое наблюдение 5-летней девочки Габриэль с необычными, непрерывно прогрессирующими аномалиями скелета, и дал патологии своё имя.[5]
Позднее выяснилось, что в действительности девочка страдала врождённой контрактурной арахнодактилией.[6]
Американский генетик Виктор Маккьюсик открыл этим синдромом новую нозологическую страницу наследственных заболеваний соединительной ткани.[7]
Симптомы[править | править код]
Фенотип больных характеризуется определённой протяжённостью: начиная от лёгких, «мягких» форм соединительнотканной дисплазии, встречающихся и в общей популяции — до случаев с угрожающими жизни системными расстройствами.[8]
Органы зрения: у половины больных диагностируется подвывих хрусталика; у лиц с выраженной миопией повышен риск отслойки сетчатки.
Мышечно-скелетная система: арахнодактилия, долихостеномелия, деформации позвоночника (сколиоз, лордоз, гиперкифоз), деформация передней стенки грудной клетки (вдавленная грудь, «куриная грудь»), гипермобильность суставов, плоская стопа, высокое готическое нёбо, недоразвитие вертлужной впадины, врождённые контрактуры локтей и пальцев, мышечная гипотония.
Сердечно-сосудистая система: пролапс митрального клапана отмечается в 80 % случаев; со временем створки клапанов утолщаются, становясь гистологически миксоматозными; дилатация корня аорты начинается с синуса Вальсальвы и прогрессирует с возрастом (у женщин отмечается более медленное прогрессирование) и в конечном итоге может приводить к расслаивающейся аневризме аорты.
Другие системы органов: у 5 % больных отмечаются спонтанные пневмотораксы; характерны стрии на коже (striae atrophicae) в областях плеч, груди, поясницы; у большинства больных наблюдается сужение нервного канала в пояснично-крестцовом отделе; нередко диагностируются кистозные образования в печени и почках, которые увеличиваются с возрастом и обычно клинически не значимы.
Многие люди с синдромом Марфана имеют высокие показатели интеллекта (выше, чем среднестатистический показатель IQ в популяции).
Диагностика[править | править код]
В рамках ревизованных Гентских критериев (2010г.) требования к диагностике синдрома Марфана различаются в зависимости от данных наследственного анамнеза.
Если семейный или наследственный анамнез не отягощен, синдром устанавливается в следующих случаях:
• при наличии подтверждённого расширения корня аорты и эктопии хрусталика;
• при наличии расширения корня аорты и подтверждённой мутации гена FBN1;
• при наличии эктопии хрусталика без вовлечения корня аорты с подтверждением мутации в гене FBN1;
• при сочетании расширения аорты и признаков системного вовлечения соединительной ткани
Лечение[править | править код]
Лечение — преимущественно симптоматическое, направлено на облегчение тех или иных проявлений заболевания. Больным необходимо проходить расширенное ежегодное медицинское обследование с обязательным участием офтальмолога, кардиолога и ортопеда.
Большинство клинических исследований поддерживают профилактическое употребление бета-адреноблокаторов с раннего возраста для предотвращения расслаивающейся аневризмы аорты. В случае выраженной дилатации корня аорты проводится его хирургическая коррекция. Показанием для операции у взрослых больных является достижение максимального диаметра корня аорты 50 мм.[9]
См. также[править | править код]
- Дисплазия соединительной ткани
- Фибриллин-1
- TGF-бета
Примечания[править | править код]
- ↑ Disease Ontology release 2019-05-13 — 2019-05-13 — 2019.
- ↑ Синдром Марфана (недоступная ссылка). Первый Московский Государственный Университет имени И.М. Сечнова (19 октября 2007). — Течение и прогноз. Дата обращения 23 августа 2012. Архивировано 14 августа 2011 года.
- ↑ Marfan Syndrome (англ.). NORD (National Organization for Rare Disorders). Дата обращения 23 сентября 2019.
- ↑ Williams E. Rare Cases, with Practical Remarks Архивная копия от 20 декабря 2016 на Wayback Machine // Transactions of the American Ophthalmological Societ. — 1875. — Vol. 2. — PP. 291—301. — PMC 1361735
- ↑ Marfan A. B. Un cas de deformation congenital des quatre membres plus prononcee aux extremities caracterisee par l’allongement des os avec un certain degre d’amincissement. // Bulletins Et Memoires De La Societe Medicale Des Hopitaux De Paris. — 1896. — Vol. 13. — PP. 220—226.
- ↑ Hecht F., Beals R. K. «New» syndrome of congenital contractural arachnodactyly originally described by Marfan in 1896. // Pediatrics. — 1972 Apr. — Vol. 49(4). — PP. 574—579. — PMID 4552107
- ↑ McKusick V. A. The cardiovascular aspects of Marfan’s syndrome: A heritable disorder of connective tissue. // Circulation[en]. — 1955 Mar. — Vol. 11(3). — PP. 321—341. — PMID 14352380
- ↑ Pyeritz R.E. Disorders of fibrillins and microfibrilogenesis: Marfan syndrome, MASS phenotype, contratural arachnoductyly and related conditions. In: Rimoin D.L., Connor J.M., Pyeritz R.E. (eds). «Principles and Practice of Medical Genetics», 3rd ed. New York: Churchill Livingstone, in press 1996.
- ↑ Goldman’s Cecil Medicine 978-1-4557-1167-3,
Литература[править | править код]
- Лисиченко О. В. Синдром Марфана. Новосибирск: Наука, 1986. 164 с.
Ссылки[править | править код]
На русском языке[править | править код]
- Группа ВКонтакте людей, страдающих синдромом Марфана
- Российское интернет-сообщество людей, страдающих синдромом Марфана
- «Дисплазия соединительной ткани» — Медицинский информационный сайт (Омская государственная медицинская академия)
- Синдром Марфана — Портал ГОУ ВПО ММА им. И. М. Сеченова
- Девушка с синдром Марфана рассказывает о своей жизни
На английском языке[править | править код]
- International Federation of Marfan Syndrome Organisations
- National Marfan Foundation (USA)
- National Institute for Health Marfan syndrome page (USA)
- Marfan Syndrome Information & Support
- Marfan Syndrome Research — Recent literature on Marfan Syndrome
- Marfan support
- Marfan Syndrome information from Seattle Children’s Hospital Heart Center
- Canadian Marfan Association
- Marfan Association UK
- Marfan de Mexico
- Norwegian Marfan Organization
- Online Mendelian Inheritance in Man
Источник