Синдром м а р с

3-М синдром – редкое аутосомно-рецессивное заболевание, сопровождающееся низкорослостью, названное в честь первых букв трех авторов (Miller, McKusick, Malvaux), впервые описавших его в 1975 г., характеризующееся лицевыми дизморфиями, пре- и постнатальной гипоплазией и рентгенологическими изменениями в костях (утончение длинных трубчатых костей и укорочение в переднезаднем направлении тел позвонков). Эндокринный статус и половое развитие у женщин нормальные, у мужчин может быть дисфункция гонад, частичное или полное бесплодие, которое обусловлено повышенным уровнем ЛГ, ФСГ, уменьшением размеров testis, и нарушением продукции спермы. В мировой литературе описано всего около 50 клинических случаев этого редкого заболевания в различных популяциях мира, частота гена неизвестна. Ген CUL7 (Куллин7), вызывающий это заболевание, был картирован и идентифицирован сравнительно недавно группой ученых из нескольких стран мира. Описано 25 различных мутаций в гене CUL7 у больных из 29 семей из различных стран мира (Тунис, Марокко, Франция, Алжир, Сирия, Поругалия, Германия, Шри-Ланка, Турция, Германия, Австрия, Италия, Суринам, Индия и Бразилия) разной этнической принадлежности.
| Симптомы 3-М синдрома: Для якутского синдрома низкорослости (ЯСН)характерны:пренатальная и постнатальная гипоплазия, лицевые дизморфии, гидроцефальная голова, широкая грудная клетка, мышечная гипотония, гиперлордоз, большой живот, брахидактилия, выступающие пятки и нормальный интеллект без эндокринных нарушений. слабая выраженность характерных рентгенологических признаков. Дистресс-синдром при рождении (42% — тяжелая асфиксия, 26 % — требуется механическая вентиляция, 12 % — гибель в ранний неонатальный период) А — маленький вес, низкий рост, лицевые дизморфии (гипоплазия средней трети лица, выступающий лоб, запавшее переносье, длинный фильтр), короткая шея, брахидактилия, большой живот, мышечная гипотония, микромелия кистей и стоп, выступающие пятки. В — девочка 5 лет: поясничный лордоз, гидроцефальная форма головы, деформация грудины, большой живот. С — мужчина 41 год: короткая шея, короткая и широкая грудина, короткая грудная клетка, пропорциональная низкорослость. D— сибсы, брат и сестра, у обоих признаки заболевания. Симптомы описанные для других популяций: Пропорциональная выраженная низкорослость (внутриутробная задержка роста, низкий вес при рождении, относительно большая голова, выраженная задержка роста в возрасте до 1 года). Характерное лицо (gloomy face): треугольный овал, широкий лоб, гипоплазия скуловых костей, запавшее переносье, мясистый нос, длинный фильтр, полные губы, выступающий подбородок. К другим нелицевым признакам относятся короткая широкая шея, выступающая трапецевидная мышца, квадратные плечи, короткая грудная клетка, деформации грудной клетки, гиперлордоз, гиперподвижность суставов, укорочение 5 пальцев и выступающие пятки. Интелект в пределах нормы. К характерным рентгенологическим признакам относится утончение длинных трубчатых костей, тонкие ребра, короткие в переднезаднем направлении позвонки и таз уменьшенных размеров. |
Дифференциальная диагностика
бывает иногда затрудена только с синдромом Сильвера-Рассела. При этом основными диагностическими критериями, позволяющими дифференцировать 3М-синдром являются характерные рентгенологические особенности, которых нет при СРС и аутосомно-рецессивный тип наследования. С другими синдромами пропорциональной карликовости: синдромами Дубовица, Блума, Mulibrey nanism и фетальным алкогольным синдромом дифференциальная диагностика больших трудностей не представляет.
Лечение: в ряде случаев гормональная GH-терапия в высокой дозе (4U1-1), начатая в предпубертатном возрасте при 3-М синдроме, может быть весьма эффективной.
Этноспецифические особенности 3-М синдрома у якутов, или «Якутский синдром низкорослости». «Якутский синдром низкорослости» (ЯСН), с таким названием описанный в 2007 году директором МИП «Генодиагностика» Н.Р. Максимовой вместе с соавторами синдром был включен в каталог генов OMIM как альтернативный известному «синдрому 3-М» (OMIM 273750). Основание для объединения этих двух синдромов под одним номером каталога – один и тот же ген CUL7 (Куллин 7), но с различными мутациями. У якутских больных синдром 3-М вызывается единственной, не описанной ранее мутацией в гене CUL7. Распространенность якутского синдрома низкорослости (ЯСН) составляет 1:7800 или 12,72 на 100 тыс. населения, среди детей якутской национальности — 36,7 на 100 тыс. чел. Наибольшая частота ЯСН встречается в Ленском, Оленекском, Усть-Майском, Амгинском, Намском, Сунтарском улусах..
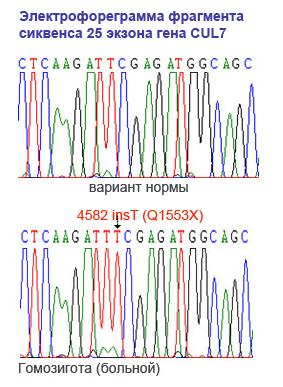 | Проведено молекулярно-генетическое изучение 37 якутских семей с аутосомно-рецессивной наследственной низкорослостью в Республике Саха (Якутия). Скрининг полного гаплоидного генома выявил генетическое сцепление с регионом 6р21,1 с максимальным lod-баллом 24,6 в локусе D6S282. Картирование гена позволило сузить кандидатный регион до 1,5 млн п. н. в области гена CUL7, ответственного за развитие синдрома 3-М. Затем был проведен мутационный анализ гена CUL7в этих семьях и идентифицирована новая нонсенс мутация 4582insT в гене CUL7 y всех 43 пациентов. |
У всех обследованных 43 больных рост родителей средний, в кровном родстве не состоят, беременность протекала в основном без осложнений (в 87% случаев роды произошли в срок). У 18 детей (41,9%) была асфиксия в родах и 11 больным (25,6%) понадобились реанимационные мероприятия в родильном зале. В 5 семьях (11,6 %) дети со схожими клиническими проявлениями, без видимых пороков развития умерли сразу же после рождения. Интеллектуальное развитие больных детей соответствовало возрасту, в половом развитии отставания не выявлено. Частота данного заболевания необычайно высока в якутской популяции, что, вероятно, обусловлено эффектом основателя. Проведенное впервые у якутов клинико-генеалогическое изучение ЯСН (3-М синдрома) показало, что для якутских больных характерны те же признаки, что и для описанных ранее в литературе клинических случаев 3-М синдрома в других популяциях мира (Miller et al., 1975; Spranger et al., 1976; Garcia-Cruz, Cantu, 1979; Cantu et al., 1981; Winter et al., 1984; Hennekam et al., 1987; von Goethem, Malvaux, 1987), за исключением дистресс-синдрома в периоде новорожденности почти у половины (41,9%) больных.
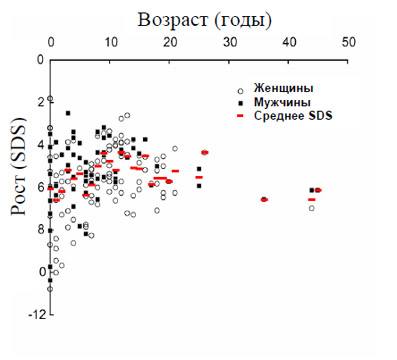 | SDS (стандартное отклонение) роста 43 больных с ЯСН при рождении и в различные возрастные периоды жизни |
Патогномоничные рентгенологические признаки для данного синдрома почти не характерны для якутских больных (тонкие длинные трубчатые кости и ребра наблюдались только у одного, высокие позвонки — у 4 больных). Выявлены расширенные метафизы у 11 больных, закрытая спинно-мозговая грыжа у 9 больных. При анализе 37 родословных в 11 случаях прослеживается аутосомно-рецессивный типнаследования: в семье было 2 и более пораженных сибсов или имелся родственник с похожим заболеванием.
Разработанная нами методика ДНК-диагностики 3-М синдрома в якутской популяции позволяет проводить дородовую молекулярно-генетическую диагностику в отягощенных семьях, а выявление гетерозиготного носительства мутации 4582insT в гене CUL7 является эффективным методом профилактики моногенной патологии в якутской популяции. Оформлен патент РФ на «Способ ДНК-диагностики 3-М синдрома в якутской популяции», Якутский синдром с низкорослостью внесен в Базу данных OMIM как синоним 3-М синдрома. Разработан алгоритм пренатальной диагностики 3М-синдрома в отягощенных семьях. Распространенность среди всего якутского населения — 12,72 на 100 тыс. населения, среди детского населения — 36,7 на 100 тыс. детского населения. Клинические признаки якутских больных с 3-М синдром сходны с клиникой других больных в мире, за исключением дистресс-синдрома новорожденных и низкой частоты рентгенологической патологии. Мутация 4582insT в гене CUL7 является единственной причиной 3-М синдрома в якутской популяции. Частота гетерозиготного носительства мутации 4582insT в якутской популяции составляет 3%. Выявленный одинаковый гаплотип является гаплотипом основателя. Возраст мутации 4582insT составляет 13,7 поколений или 342,5 лет. В популяциях эвенов, юкагиров, эвенков, бурятов Бурятии, русских Томской области гетерозиготных носителей мутации 4582insT в гене CUL7 не встречается. По данным Бурцевой Т.Г. в 2010 г. выявлена частота гомозигот ЯСН у детей саха и эвенов, 1:36 и 1:121 соответственно. Частота гетерозигот у детей эвенов составила 4:121.При этом у детей юкагиров, чукчей, эвенков данная мутация не была выявлена. Это подтверждает вывод о том, что якутская популяция, относящаяся к генетически изолированным популяциям, может быть выбрана в качестве объекта для картирования и идентификации новых генов наследственных и мультифакториальных заболеваний.
Литература:
- 3-М синдром у плода: ультразвуковые, молекулярно-генетические и гистологические особенности // Максимова Н.Р., Скрябин H.A., Павлова Н.Л. и др. //Тез. научно-практ. конференции «Итоги и перспективы развития службы охраны материнства и детства в Республике Саха (Якутия). Якутск: Изд-во «ИНИТ», 2009. С.80-86.
- 3-М синдром у плода: ультразвуковые, молекулярно-генетические и гистологические особенности // Максимова Н.Р., Скрябин Н.А., Павлова H.Л. и др. // Медицинская генетика. 2008. №2. С. 42-47.
- Генетико-эпидемиологические и социально-экономические аспекты наследственной этноспецифической патологии в Якутии // Максимова Н.Р., Сухомясова A.JI., Гуринова Е.Е. и др. // Медицинская генетика. 2008. Т. 7. №10. С. 35-43.
- Идентификация новой мутации в гене CUL7 при синдроме 3-М в якутской популяции / Н. Р. Максимова [и др.] // Медицинская генетика: ежемесячный научно-практический журнал . — 2007 . — Том 6, №11. — С. 34-38
- Идентификация новой мутации в гене CUL7 при Три-М синдроме в якутской популяции // Максимова Н.Р., Хара К., Ноговицына А.Н. и др. /Тез.13 международного конгресса по приполярной медицине // Бюллетень СО РАМН. Прил. Новосибирск, 2006. С.124.
- Клиническая характеристика 3-М синдрома у 43 якутских пациентов и подходы к ДНК-диагностике в Республике Саха (Якутии) // Максимова Н.Р., Ноговицына А.Н., Николаева И.А. и др. // Медицинская генетика. 2007. №12. С.35-38.
- Клинические и молекулярно-генетические аспекты наследственого нанизма у якутов c мутацией в гене CUL7// Н.Р Максимова, А.Н.Ноговицына, А.Л.Сухомясова, Е.Е.Гуринова, С.П.Алексеева // Якутский медицинский журнал №2(18) 2007, с. 6-10.
- Молекулярно-генетическая причина З’-М синдрома в якутской популяции //Максимова Н.Р., К.Хара, Николаева И.А. и др. // Тез. межрегиональной научно-практической конференции «Молекулярно-клеточные аспекты патологии человека на Севере». Якутск: ЯНЦ СО РАМН, 2007. С. 42-43.
- Молекулярно-генетические и гистологические особенности в плаценте и легких плода с мутацией в гене CUL7 с 3-М синдромом //Максимова Н.Р., Ноговицына А.Н., Сухомясова А.Л. и др. // Тез. II межрегиональной научно-практической конференции «Экология и здоровье человека на Севере». Якутск: ЯНЦ СО РАМН, 2007. С. 38.
- Наследственные болезни у якутов. // Пузырев В.П., Максимова Н.Р. // Генетика. – 2008.– Т.44.– №10.– С.1317-1324.
- Организационные, методические и этические проблемы ДНК-диагностики моногеных заболеваний в практике медико-генетической консультации Якутии // Кононова С.К., Федорова С.А., Степанова С.К. и др.// Мед.генетика. – 2006. – Приложение 1. – С.14-17.
- Редкий 3-М синдром у якутов: клиническая и молекулярно-генетическая характеристика // Максимова Н.Р., Ноговицына А.Н., Пузырев В.П.// Сб. VIII научно-практической конференции «Генетика человека и патология». Томск: Изд-во «Печатная мануфактура», 2007. С. 162-167.
- Редкий синдром с низкорослостью с мутацией в гене CUL7 у якутов: клиническая и молекулярно-генетическая характеристика // Максимова Н.Р., Ноговицына А.Н., Сухомясова А.Л. и др. // Тез. II межрегиональной научно-практической конференции «Экология и здоровье человека на Севере». Якутск: ЯНЦ СО РАМН, 2007. С.37.
- Респираторный дистресс-синдром у новорожденных – новый клинический признак у якутских больных с 3-М синдромом? // Н. Р. Максимова, А. Н. Ноговицына, А. Л. Сухомясова, В. А. Аргунов // Педиатрия. Журнал им. Г.Н. Сперанского, №3, том 88, 2009, с. 53-57
- Способ диагностики 3-М синдрома в якутской популяции // Максимова Н.Р., Ноговицына А.Н., Сухомясова А.Л. (Россия). Пат. 2315310 Российская Федерация, МПК вОШ 33/50 С12С> 1/68. / №2006118727/15 // Бюллетень ФИПС. Москва, заявл. 30.05.2006; опубл. 20.01.2008. №2.
- Структура и разнообразие наследственной патологии в Республике Саха(Якутия).// Тарская Л.А., Зинченко Р.А., Ельчинова Г.И., Егорова А.Г. с соавт. // Генетика, 2004. Т.40. №11. С.51-62.
- Этноспецифические наследственные болезни у якутов // Максимова Н.Р., Пузырев В.П / Тез. межрегиональной научно-практической конференции «Здоровье детей на Севере». Якутск: ЯНЦ СО РАМН, 2008. С. 91-94.
- Maksimova N., Нага К., Miyashita A. et al. Novel CUL7 mutation in 49 Yakut patients with short stature syndrome / Тез. XIII международной конференции «Human Genome Meeting». Хайдерабад, Индия, 2008. P.109.
- Maksimova N., Hara K., Miyashita A. et others. Clinical, molecular and histopathological featurea of short stature syndrome with novel CUL7 Mutation in Yakutsk: new population isolate in Asia // J. Med. Genet. – 2007. – V. 44. – P.772-778.
- Maximova N.R., Нага K., Nikolaeva I. A. et al. Novel mutation in CUL7 in Yakut patients with 3-M syndrome / Тез. международной европейской конференции European Human Genetics conference. Nice, France, 2007. P. 232.
- Maximova N.R., Sukhomyasova A.L., Nogovitsina A.N. Two forms of hereditary nanisms with autosomal recessive inheritance as one of the reasons of height retardation among children in Yakut population (Russia) / Тез. международной конференции «Eleventh International Symposium». Niigata, Japan, 2004. P. 139.
- Huber C, Dias-Santagata D, Glaser A. et others// Identification of mutations in CUL7 in 3-M syndrome// Nature Genet.2005. V. 37.P.1119-1124.
- Bailey N, Pinneau SR (1952). Tables for predicting adult height from skeletal age, revised for Greulich and Pyle hand standards. J Pediatr 40:423-441.
- Brandner MF (1970). Normal values of the vertebral body and intervertebral disk index during growth. Am J Roenl-genol 110:618.
- Brandner MF. (1972). Normal values of the vertebral body and intervertebral disk index in adults. Am J Roentgenol 114:411.
- Cantu JM, Gareia-Cruz D, Sanchez-Corona J, Fragoso R, Hernandez A, Nazara-Cazorla Z (1981). 3-M slender-boned nanism. An intrauterine growth retardation syndrome. AmJDis Child 135:905-908.
- Chatelain P, de Zegher F (1999). Growth hormone treatment in children with intrauterine growth retardation and Silver-Russell syndrome. In: Growth hormone therapy in K!GS -10 years’ experience. Editors: Rarike MB, Wilton P. Heidelberg, Leipzig: Johann Ambrosius Barth Verlag.
- Curi JFJ, Vanucci RC, Grossman H, New M (1967). Elevated serum gonadotrophins in Silver’s syndrome. Am J Dis Child 114:658-661.
- Duncan PA, Hall JG, Shapiro LR, Vibert BK (1990). Three-generation dominant transmission of the Silver-Russell syndrome. Am J Med Genet 35:245-250.
- Feldmann M, Gilgenkrantz S, Parisot S, Zarini G, Marchal С (1989). 3M dwarfism: a study of two further sibs. J Med Genet 26:583-585.
- Flannery DB (1989). 3-M syndrome. Am J Meet Genet 32:252.
- Fredriks AM et at. (1998). Nederlandsc grocidiagrammen 1997 in historisch perspectief. In: De vierde landelijke groeistudie 1997. Wit JM (red). Bureau Boerhaave Com-missie. Leiden: Rijksuniversiteit Leiden, pp. 1-14.
- Fuhrmami W, Nagele E, Gugler R, Adili E (1972). Familiiirer Minderwuchs mil unproporlionieri hohen Wirbeln. Humangenetik 16:271-282.
- Garcia-Сruz D, Cantu JM (1979). Heterozygous expression in 3-M slender-boned nanism. Hum Genet 52:221-226.
- Hennekam RCM, Bijlsma JB, Spranger J (1987). Further delineation of the 3-M syndrome with review of the literature. Am J Med Genet 28:195-209.
- Hennekam RCM (1989). Comment by Raoul CM. Hennekam on the letter to the editor by Flannery. Am J Med Genet 32:253.
- Hennekam RCM, Limburg M, Pals G (1994). 3-M syndromeand intracerebral aneurysms. J Med Genet 31:898,
- Marks LJ, Bergeson PS (1977). The Silver-Russell syndrome. A case with sexual ambiguity, and a review of the literature. Am J Din Child 131:447-451.
- Miller JD, McKusick VA, Malvaux P, Temtamy S, Salinas С (1975). The 3-M syndrome: a heritable low birthweight dwarfism. Birth Defects ХГ39-47.
- Moore GE, Abu-Amero S, Wakeling E, Hilchins M, Monk D, Stanier P, Preece M (1999). The search for the gene for Silver-Russell syndrome. Actu PaediatrSuppl 433:42-48.
- Mueller RF, Buckler J, Arthur R, Bonsor G, Dear P, Walters K, Towns GM (1992), The 3-M syndrome: risk of intracerebral ancurysm? J МЫ Genet 29:425-427.
- Poznanski AK (1984). Radiologic Anlhropomelry of the hand. In: The hand in radiologic diagnosis with Gamuts and Pattern profiles. 2nd edition.Philadelphia/Londoii/Toronto/ Mexico City/ Rio de Janeiro/Sydney/Tokyo: W.B. Saunders Company
- Price SM, Stanhope R, Garrett C, Preece MA, Trembath RC (1999). The spectrum of Silver-Russell syndrome: a clinical and molecular genetic study and new diagnostic criteria. J Med Genet 36:837-842.
- Rose S et al. (1996). Overnight growth hormone concentrations are usually normal in pubertal children with idio-pathic short stature — a clinical research center study. J Clin Endocrinol Metab 81:1063- 1068.
- Saul RA, Stevenson RE, Rogers RC, Skinner SA, Prouty LA, Flannery DB (1988). Growth references from conception to adulthood. Supplement number 1, Greenwood Genetic Center, Greenwood, pp. 142-143.
- Spranger J, Opitz JM, Nourmand A (1976). A new familial mrrauterine growth retardation syndrome. The ‘3-M syndrome1. Europ J Pediatr 123:115- 124.
- Spranger J (1977). ‘New’ dwarling syndromes. New York: Liss, A.R., Inc. For The National Foundation-March of Dimes. Birth Defects OAS X1II(3B):11- 15.
- Spranger J (1989). Reply to Dr Flannery. Am J Мed Genet 32;254.
- Van Goelhem II, Malvaux P (1987). The 3-M syndrome. A heritable low birthweight dwariism. Helv Paediatr Ada 42:159-165.
- Winter RM, Baraitser M, Grant DB, Preece MA, Hall CM (1984). The 3-M syndrome. J Med Genet 21:124-128.
- Wollmann HA, Kirchner T, Enders H, Preece MA, Ranke MB (1995). Growth and symptoms in Silver-Russell syndrome: review on the basis of 386 patients.Eur j Pediatr 154:958-968.
Источник

ARS синдром – это заболевание, поражающее сухожилия и связки, которые прикрепляются к симфизу и лобковой кости. Вначале имеет воспалительный, затем — дегенеративно-дистрофический характер. Обусловлено однообразными нагрузками и повторяющимися микротравмами. Обнаруживается у спортсменов. Проявляется болями внизу живота и паховой области, усиливающимися при отведении бедра. Диагностируется на основании жалоб, анамнеза, результатов объективного осмотра, данных рентгенографии, УЗИ и МРТ. Лечение включает ограничение нагрузок, лекарственные средства, физиотерапию, хирургические вмешательства.
Общие сведения
ARS синдром – достаточно распространенное заболевание среди спортсменов. Аббревиатура ARS расшифровывается как Adduktor-Rectus-Symphysis, содержит латинские названия пораженных структур: приводящих мышц бедра, прямой мышцы живота, лонного сочленения. Синдром известен с 1958 года, первое описание принадлежит болгарскому врачу М. Банкову. Патология входит в группу миофасциальных болевых синдромов зоны таза. Чаще всего диагностируется у футболистов. Может встречаться у лиц, активно занимающихся любыми видами спорта с интенсивной нагрузкой на ноги. Существенно ограничивает возможности пациентов, может стать причиной вынужденного ухода из большого спорта.

ARS синдром
Причины
Основной причиной развития ARS синдрома является несоответствие объема физических нагрузок и способности организма к самовосстановлению, особенно – на фоне нестабильности твердых и мягкотканных структур области таза, нижней конечности. Патология провоцируется однообразными несимметричными нагрузками на бедро, нижнюю часть живота и паховую область (например, при форсированном приведении нижней конечности в момент удара по мячу). Ситуация усугубляется непродуманным режимом тренировок и преждевременным возвращением к спортивным занятиям после травмы.
Патогенез
При перегрузке сухожилий и связок возникают микроразрывы в зонах наибольшего натяжения тканей. В ответ на повреждение образуются локальные участки отека и зоны воспаления. Устойчивость сухожилия к нагрузкам снижается, условия кровообращения в пораженной зоне ухудшаются. Перечисленное ведет к появлению все большего количества микроразрывов, образованию микрорубцов и областей жирового перерождения.
К воспалительному процессу присоединяется дегенеративно-дистрофический. Развивается энтезопатия. Формируется тендинит и тендовагинит сухожилий мышц живота и бедра в сочетании с аналогичным процессом в области связок и сухожилий симфиза. Итогом становится снижение функциональных возможностей пациента с ARS синдромом, возникновение болей.
Симптомы
ARS синдром выявляется у молодых людей, активно занимающихся спортом, обычно – у профессиональных спортсменов. Пациенты предъявляют жалобы на боль в паховой области, иррадиирующую по ходу пораженных мышц. Интенсивность болевого синдрома может варьироваться от незначительной или умеренной до выраженной, существенно ограничивающей активность больного. Отмечается связь боли с определенными физическими нагрузками. При пальпации определяется локальная болезненность в проекции сухожилий. При проведении функциональных проб (приведении бедра с сопротивлением, отведении бедра, напряжении мышц живота) болевой синдром усиливается.
Осложнения
При длительно существующем ARS синдроме из-за выраженной дегенерации сухожильной ткани возрастает вероятность крупных травм (надрывов и разрывов). В ряде случаев длительное сохранение симптоматики заболевания влечет за собой вынужденное ограничение физической активности, неучастие в соревнованиях и даже отказ от спортивной карьеры. Осложнения также могут быть обусловлены медикаментозной терапией патологии – при частых блокадах с использованием глюкокортикостероидных препаратов возможно усугубление дегенеративных процессов в пораженном отделе.
Диагностика
Предварительный диагноз нередко выставляется спортивным врачом. Для постановки окончательного диагноза требуется осмотр ортопеда и проведение аппаратных исследований. Заподозрить ARS синдром позволяет характерный анамнез (интенсивные однообразные несимметричные нагрузки), жалобы на боли в паху, усиливающиеся при движениях, положительные результаты функциональных проб. Для подтверждения назначаются следующие инструментальные методики:
- Рентгенологическое исследование. При продолжительном течении ARS синдрома на рентгенограммах таза обнаруживается наличие дегенеративно-дистрофических изменений в зоне сочленения лонных костей. Возможны аналогичные поражения крестцово-подвздошных суставов.
- УЗИ лонного сочленения. В ходе сонографии оценивается состояние хрящевых и костных структур, верхних отделов мышц бедра, областей прикрепления их сухожилий (энтезов). По результатам процедуры определяется расширение симфиза, дегенерация сухожильных и мышечных волокон, особенно выраженная в зоне, прилегающей к кости.
- МРТ костей таза. Сканирование позволяет визуализировать воспаление и дегенерацию в энтезисах и прилегающих частях сухожилий, а также в области симфиза и крестцово-подвздошных сочленений.
Лечение ARS синдрома
Консервативная терапия
Лечение может осуществляться амбулаторно или в условиях отделения травматологии и ортопедии. Важным условием успешной терапии является исключение интенсивных физических нагрузок. Больным ARS синдромом рекомендуют временно прекратить тренировки. На ранних стадиях используют медикаментозную терапию, физиотерапию. Назначают НПВС общего действия, в пораженную зону вводят кортикостероидные средства. Применяют следующие физиотерапевтические методы:
- лазеротерапия;
- магнитотерапия;
- токи Бернара;
- электрофорез с обезболивающими препаратами;
- массаж, ЛФК.
Показана кинезиотерапия на зону лонного сочленения и прилегающие мышцы. Наиболее эффективным консервативным методом лечения является ударно-волновая терапия.
Хирургическое лечение ARS синдрома
При неэффективности консервативных методик, частых рецидивах рекомендовано хирургическое вмешательство. В ходе операции при ARS синдроме производят частичное рассечение вовлеченных в патологический процесс мышц бедра и живота с их одномоментной пластической реконструкцией. Больному разрешают вставать на второй день. В послеоперационном периоде назначают анальгетики, антибактериальные средства.
Реабилитация
Через 3 недели при удовлетворительных данных УЗИ, свидетельствующих о достаточном восстановлении оперированных структур, начинают реабилитацию, которая включает в себя лечебную физкультуру, электромиостимуляцию, гидрокинезотерапию. Через месяц после хирургического лечения ARS синдрома разрешают бег без препятствий и ускорений. Через полтора месяца пациента допускают к тренировкам, рекомендуют постепенное увеличение нагрузки.
Прогноз и профилактика
Прогноз больных ARS синдромом можно рассматривать, как условно благоприятный. Эффективность консервативной терапии невелика, устойчивое улучшение наступает у 20-25%. Лучшие результаты отмечаются после проведения ударно-волновой терапии. После оперативного лечения болевой синдром исчезает, пациенты возвращаются к занятиям спортом в обычном режиме, однако в отдаленном периоде возможны рецидивы. Профилактические меры включают продуманный режим тренировок, постепенное увеличение спортивных нагрузок, обеспечение достаточного периода восстановления после травм.
Источник