Синдром леви код диагноза по мкб 10
Связанные заболевания и их лечение
Описания заболеваний
Стандарты мед. помощи
Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Синдром Русси-Леви.
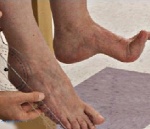
Синдром Русси-Леви
Описание
Синдром Русси. Леви — наследственное заболевание из группы мотосенсорных невропатий, по некоторым представлениям являющееся фенотипическим вариантом амиотрофии Шарко — Мари — Тута. Основные симптомы включают сенситивную атаксию, слабость и гипотрофию мышц дистальных отделов конечностей (преимущественно нижних), сухожильную арефлексию. При установлении диагноза опираются на особенности клинического симптомокомплекса и течения заболевания, семейный анамнез, результаты электронейромиографии. Пациентам показано регулярное симптоматическое лечение: антихолинэстеразные фармпрепараты, витамины, калий, АТФ, массаж, физиопроцедуры, ЛФК.
Дополнительные факты
Синдром Русси-Леви носит эпонимическое название в честь описавших его в 1926 году французских медиков невролога Леви и патолога Русси. Авторы обозначили описанный синдром как семейную арефлекторную дистазию с медленным прогрессированием. Базисными клиническими проявлениями синдрома выступают атаксия и сухожильная арефлексия, что легло в основу другого его названия — «синдром атаксии-арефлексии». Согласно международной классификации 10-го пересмотра, синдром Русси-Леви отнесен к группе наследственных мотосенсорных невропатий (НМСН). Однако среди клиницистов нет однозначного мнения, как классифицировать данную патологию. В соответствии с симптоматикой ряд специалистов в области неврологии относит синдром Русси-Леви к клинической форме, занимающей промежуточное положение между болезнью Шарко-Мари-Тута и атаксией Фридрейха, другие авторы считают его отдельным клиническим вариантом спинальной амиотрофии. Не смотря на полученные генетические доказательства последнего утверждения, многие неврологи до сих пор выделяют синдром Русси-Леви как отдельную нозологию.
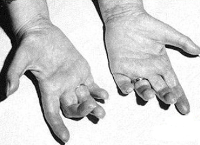
Синдром Русси-Леви
Причины
Семейный характер заболевания указывает на его наследственный генез. Ранее было установлено, что невропатия Русси-Леви встречается в семьях с повторными случаями болезни Шарко-Мари-Тута. Молекулярно-генетические исследования последних лет выявили, что генетическим субстратом синдрома выступают нарушения в 17-й хромосоме (локус 17р11. 2) в виде наличия дубликатов отдельных генов, точковые мутации гена MPZ — нулевого миелинового протеина. Это открытие позволяет считать синдром Русси-Леви фенотипической формой невральной амиотрофии Шарко-Мари-Тута (НМСН вариант IА).
Наследование синдрома осуществляется аутосомно-доминантным путем. Морфологически определяются дегенеративные изменения задних спинальных корешков и периферических нервных стволов в виде умеренной демиелинизации и уменьшения числа нервных волокон; дистрофические изменения пирамидных и мозжечковых трактов, проходящих в боковых столбах спинного мозга; дегенерация задних столбов; атрофия мышечных волокон скелетной мускулатуры.
Симптомы
Дебют симптоматики обычно приходится на детский возраст, иногда — пубертат, в отдельных случаях возможен у взрослых. Первоначальной жалобой выступает неловкость при ходьбе за счет снижения силы мышц голени (в большей степени перонеальной группы) и сенситивной атаксии. Мышечная слабость обуславливает затрудненное тыльное сгибание стоп, их свисание в состоянии подошвенного сгибания. Ранним признаком заболевания является выпадение сухожильных рефлексов нижних конечностей (коленных, ахилловых). Со временем наблюдается арефлексия верхних конечностей, слабость мышц дистальных отделов рук и атрофии мышц кисти. Отмечаются полиневритические нарушения чувствительности: болевая и температурная гипестезия в виде «носков» и «перчаток», расстройство глубоких видов (мышечно-суставного, вибрационного восприятия). Последнее обуславливает существование сенситивной атаксии.
Тремор.
Диагностика
Диагноз базируется на клинической картине заболевания и ее изменении в динамике. От невральной амиотрофии невропатию Русси-Леви отличает меньшая выраженность мышечных гипотрофий, от болезни Фридрейха — отсутствие дизартрии и склонность к стабилизации состояния. Дифдиагностика проводится также с другими видами полиневропатий, с миопатией и миотонией, боковым амиотрофическим склерозом, болезнью Рефсума, миелопатиями. Пациентам показана консультация генетика и генеалогический анализ.
С целью уточнения диагноза невролог назначает электронейромиографию, результаты которой говорят о замедлении проведения импульсов по нервным стволам. В сложных диагностических случаях проводится биопсия мышц и/или поверхностных нервов нижних конечностей. Микроскопическое исследование мышечной ткани выявляет атрофические изменения отдельных пучков мышечных волокон. При микроскопии препаратов нерва определяется разрастание элементов шванновской оболочки, приводящее к гипертрофии нервных волокон, по своей форме напоминающей луковицу.
Лечение
В настоящее время разработана только симптоматическая терапия, базирующаяся на фармпрепаратах, облегчающих проведение нервного импульса и улучшающих обменные процессы нервной ткани. Составляющими лечения обычно выступают тиамин, пиридоксин, витамин С, цианокобаламин, оротат калия, антихолинэстеразные средства (амбенония хлорид, дистигмин, галантамин, пиридостигмин, неостигмин), АТФ. Параллельно рекомендованы занятия лечебной физкультурой, массаж, физиолечение и сегментарно-рефлекторная терапия. Деформация стоп является показанием к консультации ортопеда с подбором ортопедической обуви.
Прогноз
Синдром Русси-Леви отличается хорошим для жизни прогнозом. Медленное нарастание симптоматики с последующей стабилизацией процесса обеспечивает большинству пациентов длительную сохранность трудоспособности и негрубое ее снижение. В некоторых случаях наблюдается неуклонное прогрессирование мышечной слабости и гипотрофии, приводящее к затруднению самостоятельной ходьбы.
Основные медуслуги по стандартам лечения | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Клиники для лечения с лучшими ценами
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Источник
Утратил силу — Архив
![]()
РЦРЗ (Республиканский центр развития здравоохранения МЗ РК)
Версия: Архив — Клинические протоколы МЗ РК — 2010 (Приказ №239)
Категории МКБ:
Наследственная моторная и сенсорная невропатия (G60.0)
Общая информация
Краткое описание
Наследственные невропатии представляют собой гетерогенную группу генетически детерминированных заболеваний нервной системы, проявляющихся множественным поражением периферических нервов и различающихся типом наследования, выраженностью клинического полиморфизма, особенностями течения, характером электромионейрографических и морфогистохимических изменений.
Протокол «Наследственная моторная и сенсорная невропатия»
Коды по МКБ-10: G 60,0
— Болезнь Шарко-Мари Тута (наследственная мотосенсорная невропатия, тип I)
— Болезнь Дежерина — Сотта (наследственная мотосенсорная невропатия, тип III)
— Наследственная моторная и сенсорная невропатия, типы I-IV
— Гипертрофическая невропатия у детей
— Перонеальная мышечная атрофия (аксональный тип, гипертрофический тип)
— Синдром Руси-Леви
Мобильное приложение «MedElement»
— Профессиональные медицинские справочники. Стандарты лечения
— Коммуникация с пациентами: вопросы, отзывы, запись на прием
Скачать приложение для ANDROID / для iOS
Мобильное приложение «MedElement»
— Профессиональные медицинские справочники
— Коммуникация с пациентами: вопросы, отзывы, запись на прием
Скачать приложение для ANDROID / для iOS
Классификация
Клинико-генетическая классификация наследственных мотосенсорных невропатий
Аутосомно-доминантная наследственная мотосенсорная невропатия, тип I:
Тип I А:
— обусловленная дупликацией 17 р 11.2;
— обусловленная точковыми мутациями гена миелина РМР-22.
Тип I В: обусловленная точковыми мутациями гена миелина Ро.
Тип I С: с неидентифицированным генетическим дефектом.
Аутосомно-рецессивная наследственная мотосенсорная невропатия, тип I: с неидентифицированным генетическим дефектом.
Аутосомно-доминантная наследственная мотосенсорная невропатия, тип II: с неидентифицированным генетическим дефектом.
Аутосомно-рецессивная наследственная мотосенсорная невропатия, тип II: с неидентифицированным генетическим дефектом.
Наследственная мотосенсорная невропатия, тип III:
— обусловленная дупликацией 17 р 11.2;
— обусловленная точковыми мутациями гена миелина РМР-22;
— обусловленная точковыми мутациями гена миелина Ро.
Х- сцепленная наследственная мотосенсорная невропатия:
— обусловленная точковыми мутациями и делециями коннексина 32;
— с неидентифицированным генетическим дефектом.
Сложные формы наследственных мотосенсорных невропатий:
— с пирамидными симптомами;
— с атрофией зрительных нервов;
— с атрофией зрительных нервов и глухотой;
— с пигментной ретинопатией;
— с кератодермией ладоней и стоп.
Диагностика
Диагностические критерии
Жалобы и анамнез: мышечная утомляемость, атрофии, боли в дистальных отделах конечностей по «полиневритическому типу», костные деформации, нарушение походки, задержка моторного развития, прогрессирующее течение.
В анамнезе: при болезни Шарко-Мари — аутосомно-рецессивный тип наследования, дебют заболевания в первое десятилетие жизни.
При болезни Дежерина-Сотта (наследственной мотосенсорной невропатии, тип III): аутосомно-рецессивный тип наследования, дебют в первые два года жизни, задержка моторного развития, слабость и атрофии дистальных отделов конечностей, по мере прогрессирования — поражение проксимальных отделов, атаксия, быстропрогрессирующее течение.
Для наследственной мотосенсорной невропатии, тип I характерен аутосомно-доминантный тип наследования, дебют заболевания преимущественно в 1-2 десятилетие жизни, медленно-прогрессирующий характер течения.
Для наследственной мотосенсорной невропатии, тип II — характерен аутосомно-доминантный тип наследования, дебют во второй декаде жизни, отсутствие либо незначительно выраженные атрофии дистальных отделов конечностей, относительно доброкачественное течение.
Для наследственной мотосенсорной невропатии, тип II — характерен аутосомно-рецессивный тип наследования, дебют заболевания в раннем возрасте, выраженные атрофии и слабость дистальных отделов конечностей, деформации кистей и стоп, быстропрогрессирующий характер течения.
Физикальное обследование: неврологический статус — боли (ноющие, стреляющие, крампи) в дистальных отделах конечностей на фоне гипостезии, с постепенным присоединением слабости, атрофии в дистальных отделах, снижение и утрата ахилловых рефлексов по мере прогрессирования — карпо-радиальных; нарушение походки по типу «петушиной», степпаж.
Атрофии возникают в дистальных отделах с последующей (при прогрессировании) атрофией проксимальных отделов конечностей. Деформированные стопы по типу «клюшки» или «фридрейховской» стопы в виде полой стопы с высоким сводом, экстензией основных фаланг. При дебюте заболевания в детском возрасте — задержка моторного развития.
Лабораторные исследования: общий анализ крови и мочи без патологии.
Инструментальные исследования
ЭМГ различна: для наследственной мотосенсорной невропатии, тип I характерно снижение скорости проведения импульса по моторным и сенсорным волокнам.
Для болезни Шарко-Мари снижение скорости проведения импульса по моторным и сенсорным волокнам.
Для болезни Дежерина-Сотта значительное снижение (менее 12 м/с) скорости проведения импульса по периферическим нервам.
Для наследственной мотосенсорной невропатии, тип II (аутосомно-рецессивная форма) на ЭМГ характерно значительное снижение скорости проведения импульса по периферическим нервам (менее 38 м/с).
Для наследственной мотосенсорной невропатии, тип II (аутосомно-доминантная форма) умеренное снижение скорости проведения импульса по периферическим нервам.
Показания для консультации специалистов:
1. Психолог.
2. Логопед.
3. Ортопед.
4. Генетик.
5. Протезист.
Минимум обследования при направлении в стационар:
1. Общий анализ крови и мочи.
2. АЛТ.
3. АСТ.
4. Кал на яйца глист.
Основные диагностические мероприятия:
1. Общий анализ крови.
2. Общий анализ мочи.
3. ЭМГ.
4. Логопед.
5. Психолог.
6. Окулист.
7. Протезист.
8. Ортопед.
Дополнительные диагностические мероприятия:
1. ЭКГ.
2. Кардиолог.
Дифференциальный диагноз
Дифференциально-диагностические признаки аутосомно-доминантных наследственных мотосенсорных невропатий
Признак | Аутосомно-доминантная наследственная мотосенсорная невропатия | |
Тип I | Тип II | |
Дебют заболевания | Чаще первая декада жизни (60% больных) | Чаще вторая декада жизни (70% больных) |
Мышечная слабость | Прогрессирует с возрастом | Может быть не выражена |
Деформация стоп | Наблюдается в 2/3 случаях | Наблюдается в 1/3 случаях |
Сухожильные рефлексы | Резкое снижение пяточных рефлексов | Возможно снижение пяточных рефлексов |
Чувствительные расстройства | Постоянны | Возможны |
Тремор кистей | Часто | Редко |
Скорость проведения импульса по срединному нерву | Менее 30 м/с | Более 40 м/с |
Дифференциально-диагностические признаки наследственных нервно-мышечных заболеваний
Признак | Невральная амиотрофия | Спинальная амиотрофия | Миопатии |
Дебют заболевания | Первая декада жизни | Первая декада жизни | Первая декада жизни |
Мышечная слабость | Прогрессирует с возрастом | Прогрессирует с возрастом | Прогрессирует с возрастом |
Чувствительные расстройства | Постоянны | Не характерны | Не характерны |
Деформация стоп | Наблюдается в 2/3 случаях | Не характерны | Не характерны |
Сухожильные расстройства | Резкое снижение пяточных рефлексов | Гипо-, арефлексия | Гипо-, арефлексия |
Патология сердца | Не характерна | Не характерна | Миокардиодистрофия |
ЭМГ | I тип, характерно снижение скорости проведения импульса по моторным и сенсорным волокнам | II тип, денервационный характер изменений | Первично-мышечный характер изменений |
Лечение
Тактика лечения
Цели консервативного лечения:
1. Коррекция двигательных нарушений.
2. Коррекция нервно-психического статуса пациентов.
3. Обеспечить больному социальную адаптацию.
4. Определить форму невральной амиотрофии и обеспечить адекватное лечение.
Немедикаментозное лечение:
1. Физиотерапия.
2. Лечебная физкультура.
3. Кондуктивная педагогика.
4. Массаж.
5. Ортопедическая коррекция.
Медикаментозное лечение:
1. Антиоксидатная терапия:
— никотинамид 10-20 мг/сут.;
— аевит 1 кап./сут.
2. Общеукрепляющая терапия: витамины группы В, фолиевая кислота, препараты магния.
3. Аминокислоты: церебролизин, метионин, глутаминовая кислота.
4. Препараты, влияющие на тканевой метаболизм: рибоксин, карнитин, коэнзим Q, убихинон.
5. Препараты, улучшающие периферическое кровообращение: трентал, теоникол.
6. Антихолинэстеразные препараты: прозерин, нейромидин.
Профилактические мероприятия: полноценное белковое питание с ограничением углеводов, жиров. Санация очагов хронической инфекции. Профилактика вирусных и бактериальных инфекций.
Дальнейшее ведение: диспансерное наблюдение невропатолога по месту жительства, оформление пособия по инвалидности.
Основные медикаменты:
1. L- карнитин, таблетки
2. Аевит, капсулы
3. Глутаминовая кислота, таблетки 0,5
4. Карнитина хлорид, флакон 20 % — 100
5. Магне В6, таблетки
6. Никотинамид, ампула 1% -1,0; 2,5% — 1,0
7. Никотинамид, таблетки 0,005
8. Пентоксифиллин (трентал), таблетки 0,1
9. Пиридоксин гидрохлорид, ампулы, 1 мл 5%
10. Прозерин, ампулы 0,05% 1 мл
11. Тиамин хлорид, ампулы 5% 1 мл
12. Фолиевая кислота, таблетки 0,001
13. Церебролизин, ампулы 1 мл
14. Цианокобаламин, ампулы 1 мл 200 мкг и 500 мкг
Дополнительные медикаменты:
1. Дибазол, таблетки 0,02, 0,005
2. Коэнзим Q
3. Ксантинола никотинат (теоникол), таблетки 0,15
4. Метионин, таблетки 0,25
5. Нейромидин, таблетки 20 мг
6. Нейромультивит, таблетки
7. Рибоксин, таблетки 0,2
Индикаторы эффективности лечения:
— улучшение самочувствия;
— уменьшение мышечной слабости;
— повышение эмоционального и психического тонуса;
— увеличение объема активных движений;
— уменьшение выраженности чувствительных расстройств.
Госпитализация
Показания к плановой госпитализации: прогрессирующая мышечная слабость, атрофии мышц, деформации конечностей, ограничение активных движений, мышечная гипотония, нарушение походки, расстройства чувствительности, боли, парестезии в конечностях.
Информация
Источники и литература
- Протоколы диагностики и лечения заболеваний МЗ РК (Приказ №239 от 07.04.2010)
- Петрухин А.С. Неврология детского возраста, Москва 2004
Наследственные болезни нервной системы. Под редакцией Ю.Е. Вельтищева, П.А. Темина, Москва 1998 г.
Неврология. Под редакцией М. Самуэльса, Москва 1997 г.
- Петрухин А.С. Неврология детского возраста, Москва 2004
Информация
Список разработчиков:
№ | Разработчик | Место работы | Должность |
1. | Мухамбетова Гульнара Амерзаевна | КазНМУ, кафедра нервных болезней | Ассистент, кандидат медицинских наук |
2. | Кадыржанова Галия Баекеновна | РДКБ «Аксай», психоневрологическое отделение №3 | Заведующая отделением |
3. | Серова Татьяна Константиновна | РДКБ «Аксай», психоневрологическое отделение №1 | Заведующая отделением |
4. | Балбаева Аиым Сергазиевна | РДКБ «Аксай», психоневрологическое отделение №3 | Врач-ординатор |
Прикреплённые файлы
Внимание!
Если вы не являетесь медицинским специалистом:
- Занимаясь самолечением, вы можете нанести непоправимый вред своему здоровью.
- Информация, размещенная на сайте MedElement и в мобильных приложениях «MedElement (МедЭлемент)», «Lekar Pro»,
«Dariger Pro», «Заболевания: справочник терапевта», не может и не должна заменять очную консультацию врача.
Обязательно
обращайтесь в медицинские учреждения при наличии каких-либо заболеваний или беспокоящих вас симптомов.
- Выбор лекарственных средств и их дозировки, должен быть оговорен со специалистом. Только врач может
назначить
нужное лекарство и его дозировку с учетом заболевания и состояния организма больного.
- Сайт MedElement и мобильные приложения «MedElement (МедЭлемент)», «Lekar Pro»,
«Dariger Pro», «Заболевания: справочник терапевта» являются исключительно информационно-справочными ресурсами.
Информация, размещенная на данном
сайте, не должна использоваться для самовольного изменения предписаний врача.
- Редакция MedElement не несет ответственности за какой-либо ущерб здоровью или материальный ущерб, возникший
в
результате использования данного сайта.
Источник