Синдром леопарда что это такое

Текст работы размещён без изображений и формул.
Полная версия работы доступна во вкладке «Файлы работы» в формате PDF
Синдром множественного лентиго (множественные пигментные пятна) — это полиорганный наследственный дерматоз, который по начальным буквам его составляющих называют синдром ЛЕОПАРДа (LEOPARD-синдром). Его основными наружными проявлениями являются лентиго (темно-коричневые пятна).
Клиническая картина.
В первые месяцы жизни на теле ребенка появляются пигментные пятна типа веснушек. Дети отстают в умственном и физическом развитии. В возрасте 2-5 лет появляется и прогрессирует снижение слуха, вплоть до полной его потери. В старшем возрасте основная проблема-патология сердца.
Случай редкого наследственного заболевания в практике врача-генетика.
Синдром LEOPARD (синдром множественных лентиго, ОМIМ: 151100) редкое наследственное заболевание, популяционная частота которого неизвестна. Был выделен Робертом Горлином с соавторами в 1969 году. Название синдрома является аббревиатурой главных его компонентов, соответствующих основным фенотипическим и клиническим проявлениям:
Lentigines — лентиго;
Electrocardiographic conduction abnormalities — электрокардиографические нарушения проводимости;
Ocular hypertelorism — глазной гипертелоризм;
Pulmonary stenosis — стеноз легочной артерии;
Abnormalities of genitalia — аномалии половых органов;
Retardation of growth — задержка роста;
Deafness sensorineural — нейросенсорная глухота.
Заболевание наследуется по аутосомно-доминантному типу, характеризуется высокой пенетрантностью и различной степенью экспрессивности даже в пределах одной семьи.
У большинства больных (до 90%) находят мутации гена PTNP11, локализованного в сегменте 12q24.1. кодирующего белок тирозин-фосфотазу SНS-2. Примечательно то, что данный генный дефект описан у 40% пациентов с синдромом Нунан. основные фенотипические и клинические проявления которого пересекаются с СЛ: сходные лицевые дизморфии, низкорослость, кардиопатология, аномалии половых органов и задержка полового развития, нейросенсорная тугоухость. В связи с этим был предложен еще один синоним СЛ -синдром Нунан с множественным лентиго (СЛ/СНМЛ).
С момента первого описания СЛ/СНМЛ до настоящего времени известно лишь о немногим более 100 больных. Поэтому каждый новый случай этого редкого наследственного заболевания представляет определенный научный интерес.
Учеными была обследована беременная К., 1988 года рождения. В Республиканский специализированный
центр медицинской генетики и диагностики была направлена впервые в 23—24 недели беременности. Беременность 2 брак повторный, в анамнезе рождение ребенка со стенозом легочной артерии.
При осмотре беременной врач-генетик обратил внимание на множественные диффузные лентиго темно-коричневого цвета диаметром 1—5 мм на лице, шее и теле. Кроме того, у женщины имелась легкая задержка роста, птоз, широкий плоский нос, полные губы, легкая лопоухость, короткая шея, асимметричные выступающие лопатки, кифосколиоз. Отдельные более светлые пятна на ладонях. Из гинекологического анамнеза известно о задержке полового развития — mensis с 16 лет.
Врачом-генетиком осмотрена сестра беременной 2001 года рождения. Отмечены множественные лентиго на лице, шее, туловище и ладонях. Имеется низкорослость, изменения ЭКГ — нарушение сердечной проводимости и задержка полового развития.
При анализе родословной стало известно, что вышеперечисленные фенотипические признаки характерны и для брата беременной К. (сестра и брат от разных браков их матери)
Кроме того, у брата имеется стеноз легочной артерии. По совокупности клинических и фенотипических признаков был предположен диагноз: СЛ/СНМЛ. Было проведено исследование образцов ДНК крови беременной К. и ее сестры методом прямого автоматического секвенирования кодирующей последовательности экзонов 7, 12 и 13 гена РТNР11. В экзоне 7 у обеих обследуемых обнаружена идентичная мутация с.836А>G (р.Туr279Суs) в гетерозиготном состоянии. Диагноз СЛ/СНМЛ был подтвержден молекулярно-генетическим методом.
Ранее нами упоминалось, что впервые беременная К. обратилась в Центр только в 23—24 недели. Поскольку спектр генетических обследований в этом сроке весьма ограничен, беременной было выполнено только УЗИ плода с синдромологическим анализом. На момент осмотра грубых аномалий развития не выявлено, однако у плода было отмечено увеличение шейной складки. Имелось многоводие.
Беременность завершилась физиологическими родами в срок. Девочка, родилась с массой 3250 г. До 3 мес. развивается соответственно возрасту. Участковым врачом-педиатром при плановом осмотре были отмечены шумы в сердце. В результате УЗИ диагностирован субаортальный стеноз.
Ребенок осмотрен врачом-педиатром-генетиком в 3,5 месяца. Психофизическое развитие соответствует возрасту. Отмечен орбитальный гипертелоризм, короткая шея с избыточной кожей, широко расставленные соски. Пигментные пятна отсутствуют. Аналогичные признаки характерны и для старшего сибса (возраст 3,5 года). Кроме того, у старшей девочки отмечены птоз и выступающие лопатки.
Принимая во внимание особенности фенотипа у детей и наличие у них сердечной патологии, выставлен клинический диагноз: СЛ/СНМЛ. Отсутствие пигментных пятен (одного из наиболее часто встречаемых признаков) у обоих детей на момент осмотра может быть обусловлено вариабельностью как самого признака — в отдельных случаях заболевания отмечено отсутствие лентиго, так и вариабельностью периода манифестации пигментных пятен врожденные и появляющиеся в детстве, вплоть до полового созревания.
Список литературы.
Джонс Кеннет Л. Наследственные синдромы по Дэвиду Смиту. Атлас-справочник. М.: Практика, 2011.-1024 с.
Gorlin R.J. Anderson R.C. Blaw M. Multiple Ientigines. syndrome// Am. J. Dis. Child.-1969.- Vol/ 117 №6 – P. 652-662.
Digilio M.C.. Conti E., Sartozy A. et al. Grouping of multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene // Am. J. Hum. Genet.- 2002.- Vol. 71 №2 P.389-394
Источник
Синдром L.E.O.P.A.R.D
Гаджимурадов М.Н., Хачалов Г.Б., Ганиев К.Д.
Республиканский кожно-венерологический диспансер МЗ РД, г. Махачкала
Синдром L.E.O.P.A.R.D (син.: синдром множественных лентигинозных пятен, врожденный системный лентигиноз) – редко встречающееся заболевание, которое наследуется по аутосомно-доминантному типу. Кожные проявления сочетаются с комплексом дисморфогенетических расстройств.
Название синдрома L.E.O.P.A.R.D. дано с учетом начальных букв английских обозначений основных симптомов этого синдрома :
1) Lentigines – лентиго-пигментарные, распространенные, плоские или слегка возвышающиеся пятна желто-коричневого или почти черного цвета диаметра 1,5 – 3,0 см, кое-где сливающиеся, располагающиеся диссеминированно по всему кожному покрову (туловище, конечности), однако лицо обычно не поражается.
2) ECG abnormalities – изменение ЭКГ (одно- или двусторонняя гипертрофия, удлинение интервала P-Q, отклонение оси сердца, нарушения в проводящей системе сердца);
3) Ocular changes – глазной гипертелоризм (широко расставленные глаза);
4) Pulmonary stenosis – сменоз легочной артерии и другие пороки сердца (наряду с функциональными шумами в области сердца у отдельных больных обнаруживали изменения со стороны клапанов многих артерий, аорты, митральных клапанов);
5) Abnormalities of genitalia – аномалии развития половых органов (неопущение, дисплозия яичек, запоздалое появление mensis);
6) Retardation – зфдержка роста и развития;
7) Deafness – нейросенсорная глухота.
Наиболее часто (в 84 % случаях) наблюдается лентиго, в 66 % — изменения ЭКГ, в 38 % — стеноз легочной артерии и другие пороки сердца, в 33 % — глухота или тугоухость, карликовый рост (аномалии скелета) в 10 – 25 %, патология гениталий в 13 %, глазной гипертелоризм в 9 % случаев.
В литературе описано около 40 случаев этого заболевания. Характеризуется генерализованными рассеянными по всему кожному покрову (кроме лица) лентигинозными высыпаниями желто-коричневого или почти черного цвета (lentiginus), иногда сливающимися. Пятна появляются уже при рождении или в первые годы жизни независимо от воздействия солнечных лучей (в отличие от веснушек) и сохраняются постоянно. Высыпания обычно сочетаются с изменениями ЭКГ (отклонение оси сердца, гипертрофия и др.), глазным гипертелоризмом (широко расставленные глаза), стенозом легочной артерии, аномалией развития половых органов, задержкой роста и развития, нейросенсорной глухотой. Наличие 2 – 3 симптомов данного синдромапозволяет выставить диагноз.
Нами наблюдалась женщина 31 года с диссеменированными папулезными и лентигинозными элементами. В 9-месячном возрасте появились папулезные элементы на конечностях, которые регрессировали летом, обострялись зимой. На этом фоне , в 3-летнем возрасте, стали появляться единичные пигментные пятна на спине, которые к 25 годам распространились по всему кожному покрову. Пациентка отставала в росте от сверстников. Менструации начались в 13 лет. Страдает железодефицитной анемией (периодически гемоглобин падает до 52 г/л), хроническим гастритом, гипотонией (А/Д – 90/60 мм рт. ст.).
По всему кожному покрову и волосистой части головы располагаются папулы розово-красного цвета с серебристо-белыми чешуйками. При поскабливании возникает триада феноменов: стеаринового пятна, терминальной пленки, кровяной росы. Также на туловище, верхних и нижних конечностях (кроме лица) рассеяны плоские желто-коричневые пятна (0,5 – 1,5 см) с тенденцией к слиянию. У пациентки определяется глазной гипертелоризм.
Лабораторные исследования: общий анализ мочи без патологических изменений. В клиническом анализе крови гемоглобин 104 г/л. Рентгенография легких и ЭКГ в норме. Половые органы сформированы правильно.
Диагноз: L.E.O. P.A.R.D. – синдром, псориаз.
Пациентке проведён курс терапии: глюконат кальция 1 таб. х 3 раза в день, супрастин 1 таб. х 2 раза в день; 30 % тиосульфат натрия внутривенно по 10 мл 1 раз в день (№ 10). Наружно – 2 % салициловая мазь. На фоне лечения большая часть папулезных элементов запала, поблекла и регрессировала, однако лентигинозные пятна изменений не претерпели.
Источник
Синдром множественного лентиго (множественные пигментные пятна) — это полиорганный наследственный дерматоз, который по начальным буквам его составляющих называют синдром ЛЕОПАРДа (LEOPARD-синдром). Его основными наружными проявлениями являются генерализованные лентиго (темно-коричневые пятна).
Ниже перечислены 7 основных составляющих:
L: Lentigines (лентиго)
Е: EKG abnormalities (отклонения на ЭКГ)
О: Ocular hypertelorism (гипертелоризм глаз)
Р: Pulmonary stenosis (стеноз легочной артерии)
A: Abnormal genitalia (аномалии половых органов)
R: Retardation of growth (задержка развития или карликовость)
D: Deafness (глухота)
Синонимы: синдром множественных лентиго, сердечно-кожный синдром, обструктивная гипертрофическая кардиомиопатия, связанная с лентиго, лентигиноз, профузное лентиго, генерализованный лентикулярный меланоз, синдром профузного лентигиноза, прогрессирующий миопатический лентигиноз, синдром Мойнахана, синдром Горлина.
Эпидемиология синдрома множественного лентиго
Возраст: лентиго обычно появляются при рождении. Внекожные проявления не появляются до полового созревания, возраст клинических проявлений — 14 лет.
Пол: не имеет значения.
Распространенность: встречается редко.
Генетика: аутосомно-доминантный тип наследования с различной степенью выраженности. Этиология: мутация в гене PTPN11.
Анамнез синдрома множественного лентиго
Лентиго часто наследуются, и к половому созреванию число, размеры и интенсивность окраски кожных элементов увеличиваются. Однако в дальнейшем они начинают бледнеть. Внекожные патологические изменения часто проявляются только при половом созревании и включают в себя стеноз легочной артерии, обструктивную кардиомиопатию, изменения в межпредсердной перегородке, первичную легочную гипертензию, деформацию грудной клетки, кифосколиоз, гипоспадию, крипторхизм, умственную отсталость, нейросенсорную тугоухость.
Патологические изменения сердца приводят к значительному ухудшению состояния при этом синдроме.
Клиника синдрома множественного лентиго
Тип высыпаний: несколько четко ограниченных макул.
Размер: от 1 до 5 мм.
Цвет: темно-коричневый или черный.
Форма: круглая или овальная.
Локализация: лентиго сконцентрированы на лице, шее и верхней части туловища, но могут присутствовать на руках, ладонях, ступнях и на наружных половых органах. Слизистые оболочки остаются незатронутыми.
Общие проявления синдрома множественного лентиго
Скелет: отставание в росте, гипертелоризм, изменение формы грудной клетки, кифосколиоз, крыловидная лопатка. Сердечно-сосудистая система: стеноз легочной артерии и нарушение внутрисердечной проводимости.
Мочеполовая система: гипоплазия гонад, агенезия почек.
Нервная система: перцептивная тугоухость; отклонения в ЭЭГ, замедление проводимости в периферических нервах.
Гистопатология синдрома множественного лентиго: увеличение меланоцитов в базальной мембране.
Электронная микроскопия: увеличение количества/размеров меланосом в кератиноцитах.
Дифференциальная диагностика синдрома множественного лентиго. Следует учитывать другие лентигинозные синдромы, такие как синдром Пейтца-Егерса. В то время как при синдроме множественных лентиго образования в основном локализуются на лице, не затрагивая слизистые оболочки, при синдроме Пейтца-Егерса наблюдается выраженное поражение слизистой полости рта.
Течение и прогноз синдрома множественного лентиго. Наиболее серьезными являются изменения скелета, сердца и эндокринной системы. Пятна лентиго доставляют только косметические проблемы, становясь со временем темнее, больше и многочисленнее.
Лечение синдрома множественного лентиго
Пятна лентиго являются доброкачественными образованиями, лечение не требуется. Пятна могут быть скрыты косметическими средствами и, в конце концов, со временем посветлеть. Для ускорения этого процесса можно применить перекись водорода, салицилаты, гидрохинон, третинон, жидкий азот или лазер. Лечение должно применяться без чрезмерного обесцвечивания.
Наиболее важными при синдроме ЛЕОПАРДа являются изменения скелета, сердца и эндокринной системы. Несмотря на то, что внекожные изменения не проявляются до полового созревания, пациенты при синдроме ЛЕОПАРДа нуждаются в наблюдении и лечении стеноза легочной артерии, обструктивной кардиомиопатии, дефектов межпредсердной перегородки, первичной легочной гипертензии, деформации грудной клетки, кифосколиоза, крипторхизма, умственной отсталости и тугоухости.
— Читать «Пятна кофе с молоком (макулы цвета кофе с молоком, МКЦМ) — диагностика, лечение»
Оглавление темы «Дерматология»:
- Гало-невус — диагностика, лечение
- Пятнистый невус (шпилюс-невус) — диагностика, лечение
- Невус Шпитц (веретеноклеточный и эпитеолиоидный невус) — диагностика, лечение
- Эфелиды (веснушки) — диагностика, лечение
- Простое лентиго — диагностика, лечение
- Синдром Пейтца-Егерса — диагностика, лечение
- Синдром множественного лентиго (LEOPARD-синдром) — диагностика, лечение
- Пятна кофе с молоком (макулы цвета кофе с молоком, МКЦМ) — диагностика, лечение
- Монгольское пятно (наследуемый кожный меланоцитоз) — диагностика, лечение
- Невусы Ото и Ито — диагностика, лечение
Источник
Патология, наследственность и дифференциация синдрома LEOPARD
Биопсия кожи, взятой из гиперпигментированной области, обнаружила удлинение сети шиповидных клеток, множественные меланоциты и увеличение пигмента в базальных клетках.
Наследственность. На основании широкого диапазона клинических проявлений у разных больных создается впечатление, что этот синдром наследуется по аутосомно-доминантному типу с варьирующей экспрессивностью (Gorlin et al.).
Наиболее достоверными клиническими симптомами болезни являются пигментные пятна, отсутствующие обычно при рождении, но прогрессирующие впоследствии. От их присутствия явно зависит диагностика.
Диагноз. Черты, имеющие некоторое сходство с этим синдромом, встречаются при нескольких заболеваниях. Noonan и Ehmke описали несколько детей с врожденным заболеванием сердца, у которых наблюдались аномальное лицо, стеноз легочной артерии и другие внесердечные аномалии. К этой группе аномалий был приклеен ярлык «синдром Нунана». У больных с этим синдромом описывают также «тернеровскнн фенотип».
Синдром Нунана встречается как у мальчиков, так и у девочек; его симптомы имеют много общего с синдромом множественных пигментных пятен: гипертелоризм, птоз, низкий рост, стеноз легочной артерии без звука захлопывания клапана, аномальная ось QRS (чаще в S1, S2, S3), неопущение яичек, замедленное развитие вторичных половых признаков, аномалии скелета грудной клетки и вероятное ауто-сомно-доминантное наследование с неполной пенетрантностыо. У больных с синдромом Нунана могут быть случайно обнаружены крыловидные складки на шее, но у них не бывает множественных пигментных пятен и глухоты.
Связь между синдромом множественных пигментных пятен и синдромом Нунана не ясна. Возможно, оба синдрома представляют собой результат плейотропного эффекта какого-то гена с акцентом его действия на локализации, определяющей различие проявлений. Вероятно также, что они могут быть представлены аллельными мутациями. Кроме того, оба синдрома имеют определенное сходство с рубеолярной эмбриопатией: стеноз легочной артерии, аномалия осп комплекса QRS, низкий рост, задержанное половое развитие и глухота (как при синдроме множественных пигментных пятен). Ангиографически при краснушном синдроме выявлен стеноз легочной артерии, имеющий выраженное сходство с тем, что наблюдается у больных с дисплазией клапана легочной артерии.
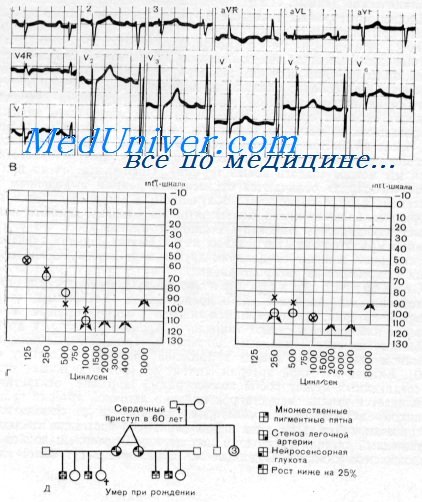
Доминантная пегость, атаксия и нейросенсорная глухота с обширными областями депигментации резко отличаются от поражения при синдроме множественных пигментных пятен. Множественные пигментные пятна также вполне отличимы от резких нарушений пигментации с областями гиперпигментации, обнаруженных при синдроме Х-сцепленной пигментной патологии с врожденной нейросепсорпой глухотой.
Лечение. Резко выраженный стеноз легочной артерии подлежит оперативному лечению при помощи методов сердечной хирургии (Gorlin et al.). Пигментные пятна можно лечить путем отшелушивания кожи (Selmanowitz et al.). Больным с глухотой могут помочь слуховые аппараты. Крипторхизм должен быть исправлен при помощи операции.
Прогноз. По-видимому, прогрессирующим симптомом этого заболевания является только увеличение числа пигментных пятен в течение первых двух десятилетий жизни. Некоторые больные с тяжелой обструкционной кардиомиопатией могут погибнуть в молодом возрасте (Somerville, Bonham-Carter), по у большинства продолжительность жизни нормальная.
Выводы: характеристика этого синдрома включает:
1) аутосомно-доминантное наследование с варьирующей экспрессивностью;
2) множественные пигментные пятна, развивающиеся после рождения;
3) дефект ЭКГ, обнаруживающий некоторые комбинации блока в системе пучка Гиса;
4) стеноз легочной артерии и/или гипертрофию миокарда;
5) гипертелоризм;
6) аномалии половых органов, включающие крипторхизм и гипоспадию;
7) соматическую и умственную отсталость;
8) крыловидные лопатки и различные малые скелетные аномалии;
9) нейросенсорную глухоту варьирующей тяжести.
— Также рекомендуем «Врожденная глухота с рецессивной пегостью»
Оглавление темы «Наследственные болезни с глухотой»:
- Глазо-зубо-костная дисплазия. Синдром Пфейффера и Сетре—Чотцена
- Синдром Варденбурга: клиника, диагностика
- Патология, наследственность и дифференциация синдрома Варденбурга
- Глухота с кожно-глазным альбинизмом
- Синдром множественных пигментных пятен. Синдром LEOPARD
- Патология, наследственность и дифференциация синдрома LEOPARD
- Врожденная глухота с рецессивной пегостью
- Х-сцепленные нарушения пигментации с врожденной глухотой
- Доминантная пегость с атаксией и глухотой
- Витилиго, мышечная атрофия с ахалазией и глухотой
Источник