Синдром ларсена по мкб 10

Рубрика МКБ-10: Q74.8
МКБ-10 / Q00-Q99 КЛАСС XVII Врожденные аномалии пороки развития, деформации и хромосомные нарушения / Q65-Q79 Врожденные аномалии пороки развития и деформации костно-мышечной системы / Q74 Другие врожденные аномалии (пороки развития) конечности(ей)
Определение и общие сведения[править]
Врожденные ложные суставы костей голени у детей
Врожденный ложный сустав (ВЛС) — врожденная патология сегмента кости в виде ее деформации, кисты или ложного сустава, развивающегося на почве нейрофиброматоза, фиброзной дисплазии или миелодисплазии.
Описаны врожденные ложные суставы ключицы, костей предплечья, однако наибольший практический интерес представляет врожденный ложный сустав голени.
ВЛС костей голени — редкое, трудно поддающееся лечению заболевание с различными клиническими проявлениями от легкой деформации до обширного дефекта кости.
Эпидемиология
ВЛС костей голени — редкая (заболеваемость среди новорожденных составляет 1 на 150 000) и крайне тяжелая ортопедическая патология детского возраста. Удельный вес врожденных ложных суставов среди врожденной патологии опорно-двигательного аппарата составляет 0,5-1,0%. Поражение кости чаще одностороннее и локализуется на границе средней и дистальной трети большеберцовой кости. В половине случаев поражаются обе кости голени. Связи с полом и стороной поражения не выявлено, как и с национальностью и расовой принадлежностью.
Классификация
Многочисленные классификации ВЛС основаны на рентгенологических и морфологических изменениях костной ткани. Наиболее известны следующие из них. Классификация Кроуфорда отражает этапы развития врожденного сустава.
• Тип 1 — дугообразная деформация большеберцовой кости с начальными признаками усиления плотности кортикальной пластинки и склероза костномозгового канала.
• Тип 2 — выраженная дугообразная деформация, склероз костномозгового канала в зоне патологии.
• Тип 3 — дугообразная деформация с кистой или начальными признаками патологического перелома.
• Тип 4 — деформация с явным патологическим переломом или ложным суставом, часто обеих костей.
Классификация Бойда имеет прогностическое значение.
• Тип 1. Дугообразная деформация большеберцовой кости сочетается с другими врожденными пороками.
• Тип 2. Деформация большеберцовой кости в виде песочных часов. Перелом обычно возникает в возрасте 2 лет. Концы кости тонкие, округлые, с выраженным склерозом; костномозговой канал облитерирован. Этот тип наиболее характерен для нейрофиброматоза I типа и имеет неблагоприятный прогноз из-за частых рецидивов патологии (вопреки лечению) в период роста ребенка.
• Тип 3. Врожденный ложный сустав развивается из костной кисты, обычно локализованной на границе средней и дистальной трети кости. Деформация больше-берцовой кости может предшествовать перелому или следовать за его развитием. Этот тип наиболее благоприятен для лечения, рецидивы крайне редки.
• Тип 4. Склероз костной ткани с полным или частичным склерозом костномозгового канала. Дугообразная деформация кости отсутствует. Постепенно развивается усталостное разрушение кости и формируется псевдоартроз. При раннем оперативном лечении прогноз благоприятен.
• Тип 5. Диспластическое изменение только малоберцовой кости. Прогноз благоприятный.
• Тип 6. Врожденный ложный сустав развивается вследствие внутрикостной фибромы или шванномы. Прогноз зависит от характера внутрикостного повреждения.
Наиболее известна классификация Эпойла.
• Тип I. Атрофический ложный сустав с истончением концов кости, возникающий в результате потери костной массы и склероза костномозгового канала.
• Тип II. Гипертрофический ложный сустав. Концы кости расширены, утолщены, костномозговой канал склерозирован.
Этиология и патогенез[править]
ВЛС костей голени у детей могут быть местным проявлением различных системных заболеваний. Эта патология может быть одним из клинических проявлений нейрофиброматоза. Проведенные рентгенологические и морфологические исследования показали, что в ряде случаев образованию псевдоартроза предшествуют патологические переломы в области очага фиброзной дисплазии считают, что, помимо нейрофиброматоза и фиброзной дисплазии, причиной врожденного ложного сустава голени может быть миелодисплазия.
Ни одна из общепринятых теорий не дает всеобъемлющего объяснения патогенеза врожденных ложных суставов и особенно локализации поражения. Доказано, что ключевая роль принадлежит межотломковой фиброзной ткани, формированию патологической надкостницы и нарушению кровоснабжения в зоне ложного сустава. Сама надкостница может формировать фиброзный тяж, приводящий к повышению давления вокруг кости, что также нарушает васкуляризацию и приводит к атрофии костной ткани. Нарушение васкуляризации может быть вторичным в результате утолщения сосудистой стенки в зоне псевдоартроза. В конечном итоге в силу различных причин на фоне нарушения дифференцировки остеобластов повышается активность остеокластов, что и приводит к расстройству ремоделирования кости.
Клинические проявления[править]
Ребенок редко рождается с уже сформировавшимся ложным суставом. Обычно в первые недели жизни обнаруживают дугообразную деформацию голени. С ростом ребенка, особенно с началом ходьбы, деформация прогрессирует, постепенно формируется патологический перелом, а к 2 годам — характерная клиническая картина заболевания.
Возможные клинические проявления сформированного врожденного ложного сустава: углообразная деформация конечности на границе средней и дистальной трети, утолщение кожного покрова вплоть до омозолелости на вершине деформации, гипотрофия мышц, разной степени укорочение голени, недоразвитие и неправильная установка стопы, контрактура в голеностопном суставе. Конечность не способна выполнять опорную функцию и патологически подвижна в дистальном отделе (подвижная форма ВЛС — врожденных ложных суставов). При тугой форме ВЛС ребенок может нагружать конечность при ходьбе.
Другие уточненные врожденные аномалии конечности(ей): Диагностика[править]
Диагноз устанавливают на основании данных клинического и рентгенологического обследования. Однако для уточнения этиологии заболевания необходимы консультации дерматолога, невролога и генетика, а также выполнение магнитно-резонансной томографии (МРТ).
Основополагающий метод диагностики ВЛС — рентгенография.
В латентную стадию ВЛС на прямой и боковой рентгенограммах определяют дугообразную деформацию в переднезадней и боковой плоскости, сужение костномозгового канала, утолщение кортикальной пластинки на вогнутой части деформации и истончение ее на выпуклой стороне. В последующем нарастают выраженность деформации и склероза костномозгового канала. На вершине искривления появляется косопоперечная линия патологического перелома. Позднее концы костей истончаются (атрофичный сустав) или утолщаются (гипертрофичный сустав). При кистозной форме на вершине деформации обнаруживают костную кисту, сужение костномозгового канала, а затем — поперечный перелом. При нейрофиброматозе деформация на рентгенограмме более выражена уже при рождении: кость имеет форму песочных часов, а иногда выявляют уже произошедший перелом или сформированный псевдоартроз. На рентгенологических признаках основаны классификации ВЛС.
МРТ позволяет детализировать картину мягкотканного компонента врожденного псевдоартроза, в том числе состояние надкостницы, окружающей ложный сустав. По ее толщине и виду сосудистых отверстий определяют степень васкуляризации (гиперваскуляризованный или гиповаскуляризованный ложный сустав), уточняют границы резекции кости.
Дифференциальный диагноз[править]
Другие уточненные врожденные аномалии конечности(ей): Лечение[править]
Лечение врожденных ложных суставов голени только оперативное.
В настоящее время наиболее широко используют три основных хирургических метода — интрамедуллярный остео-синтез с костной пластикой, компрессионно-дистракционный остеосинтез и васкуляризованную костную пластику. Последний метод наиболее эффективен.
Оперировать пациентов следует в возрасте 2-3 лет. До этого необходимо проводить консервативное лечение, направленное на поддержание врожденного ложного сустава в латентной форме. Фиксация деформированной конечности гипсовыми лонгетами, шинками из поливика, массаж мышц конечности, ЛФК препятствуют формированию типичной формы заболевания и создают наиболее благоприятные условия для последующей васкуляризованной костной пластики. Ошибка данного периода заключается в выполнении корригирующей остеотомии при дугообразной деформации голени (т.е. при латентной форме заболевания). Данное вмешательство не приводит к сращению и ускоряет формирование типичного псевдоартроза.
При лечении врожденных ложных суставов перед врачом стоят следующие задачи:
• восстановление опороспособности конечности;
• восстановление анатомической длины конечности;
• ликвидация патологической ткани;
• сохранение потенции конечности к росту;
• устранение контрактур.
Оперативное лечение показано как пациентам с впервые выявленной патологией, так и пациентам, у которых при использовании других методов лечения не удалось достичь положительного исхода. Противопоказанием к васкуляризованной костной пластике служат тяжелые сопутствующие пороки внутренних органов и отсутствие на конечности условий для сосудистой пластики.
Профилактика[править]
Прочее[править]
Аутосомно-доминантный синдром Ларсена
Определение и общие сведения
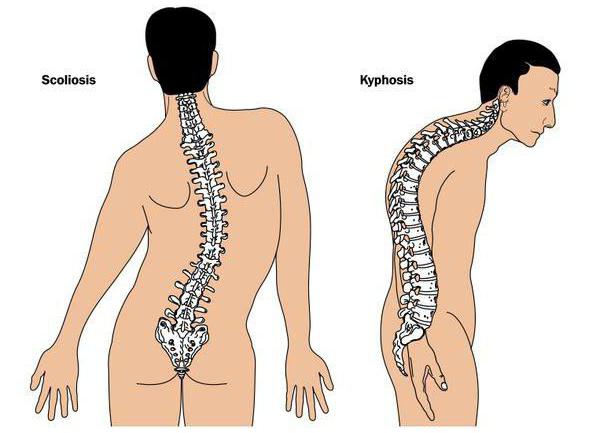
Синдром Ларсена является редкой скелетной дисплазией, характеризуется врожденным вывихом крупных суставов, деформацией стопы, дисплазией шейного отдела позвоночника, сколиозом, шпательобразными дистальными фалангами и характерными краниофациальными аномалиями, включая расщепление нёба.
Распространенность синдрома Ларсена в Европе составляет 1/250 000 новорожденных.
Синдром Ларсена передается по аутосомно-доминантному типу. Аутосомно-рецессивные случаи, описанные ранее, возможно, связаны с родительским зародышевым мозаицизмом, но, скорее всего, представляют собой рецессивные синдромы со схожей клиникой.
Этиология и патогенез
Синдром Ларсена связан с миссенс мутацией или небольшими «в рамке» делециями гена FLNB (локализуется 3p14.3), который кодирует белок цитоскелета филамин B.
Клинические проявления
Основными клиническими характеристиками синдром Ларсена являются врожденные вывихи бедра, коленного и локтевого суставов с эквиноварусная или эквиновальгусной деформаций стопы (косолапость). Другие частые проявления включают деформации позвоночника, такие как сколиоз и кифоз шейного отдела позвоночника, иногда связанные с цервикальной миелопатией и короткими, широкими, шпатель-образной формой дистальных фаланг большого пальца. Частые краниофациальные мальформации включают в себя выпуклый лоб, низкую переносицу, сплющенную и среднюю часть лица и глазной гипертелоризм. Срединное расщепление нёба, приводящее к потере слуха — также частый признак синдрома Ларсена.
Диагностика
Диагноз устанавливается на основе характерной клиники и рентгенограмм скелета, среди прочих методов — генетическое тестирование точек окостенения запястных и преплюсневых костей.
Дифференциальный диагноз
Дифференциальная диагностика включает в себя другие более тяжелые и смертельные расстройства, связанные с мутациямя гена FLNB: ателостеогенез типов I и III, бумеранг-дисплазию, а также отопталогический синдром тип I и CHST3-связанную скелетная дисплазию, хондродисплазию с дислокацией суставов типа gPAPP, Ларсен-подобный синдром тип B3GAT3, синдром Ларсена острова Реюньон и синдром Дебуке.
Лечение
Ведение пациента может включать в себя ортопедическое лечение, хирургическую коррекцию вывиха бедра и физиотерапию. Крайне важно, чтобы все дети с клиническим диагнозом синдром Ларсена шейного отдела позвоночника диагностировали как можно скорее после рождения, чтобы исключить опасную для жизни нестабильность шейного отдела позвоночника .
Прогноз
Синдром Ларсена не влияет на продолжительность жизни.
CHST3-связанная скелетная дисплазия
Синонимы: рецессивный синдром Ларсена
Рецессивный синдром Ларсена — очень редкое заболевание костей, характеризуется клинически пренатальной низкорослостью, вывихами голени, бедра или коленей, косолапостью, ограничением диапазона движения крупных суставов, прогрессирующим кифозом и иногда сколиозом. У некоторых пациентов также был описана незначительная дисплазия клапанов сердца. Интеллект, зрение и слух в норме.
Гемимелическая эпифизарная дисплазия
Определение и общие сведения
Гемимелическая эпифизарная дисплазия — редкое системное заболевание, характеризуещееся нарушением пролиферации суставного хряща с появлением дополнительных центров оссификации эпифизов, приводящих к поражению одного или нескольких из них с развитием артроза и вторичных деформаций костей скелета. Поражение при гемимелической эпифизарной дисплазии всегда одностороннее.
Впервые гемимелическая эпифизарная дисплазия под названием «тарзомегалия» была описана A. Monchet и J. Belot в 1926 г. у 18-месячного ребенка с односторонним поражением таранной кости. В дальнейшем сначала D. Trevor (1950), а затем D. Angio и соавт. (1955) описывают ряд случаев заболевания, называя его тарзоэпифизарной аклазией. В 1956 г. T. Fairbank, собрав сведения о 27 больных, дал наиболее подробное описание процесса и предложил название, используемое по настоящее время.
Заболеваемость гемимелической эпифизарной дисплазией составляет 1 случай на 1 000 000 населения.
Этиология и патогенез
Основной молекулярно-биохимический дефект, ответственный за развитие гемимелической эпифизарной дисплазии, не выявлен. Заболевание рассматривают как диспластический процесс, проявляющийся в задержке формирования субхондральной костной пластинки и продолжении костеобразования на основе закрывшейся в срок ростковой зоны суставного хряща эпифиза.
Клинические проявления
Выделяют две формы гемимелической эпифизарной дисплазии — локальную (поражение одного или нескольких эпифизов одного сустава) и распространенную (с поражением нескольких суставов).
Заболевание чаще встречается у мальчиков и может поражать любые кости скелета, но чаще всего в патологический процесс вовлекаются таранная кость и дистальные эпифизы бедренных и большеберцовых костей. Как правило, страдает только половина эпифиза (обычно медиальная), но возможно и его тотальное поражение. Одна половина эпифиза растет и развивается нормально, синостоз ее с диафизом наступает в обычные сроки. Пораженная же часть, значительно увеличиваясь в размерах, избыточно разрастается в сторону сустава, приводит к деформации зоны роста и кости в целом, развитию деформирующего артроза и нарушению биомеханики конечности, обусловливая соответствующую клиническую и рентгенологическую симптоматику.
Гемимелическая эпифизарная дисплазия может быть диагностирована по совокупности характерных клинических данных и результатов лучевых исследований.
Наибольшие изменения в суставе выявляют в период активного роста и возрастающих нагрузок на сустав. Наиболее постоянные клинические симптомы — припухлость и деформация пораженного сустава. Болевой синдром и ограничение движений возникают в более поздние сроки.
Диагностика
При рентгенологическом исследовании выявляют асимметрию эпифиза: пораженная его половина массивная, увеличенная, причудливо деформированная, неоднородной структуры с участками обызвествления. В ряде случаев обнаруживают свободно лежащие внутрисуставные тела.
Наиболее информативный метод лучевой диагностики — компьютерная томография. С помощью КТ выявляют точные взаимоотношения между костями, мягкими тканями и патологическими разрастаниями в аксиальной плоскости. Это исследование дает точную информацию об оссифицированной части образования и взаимоотношении ее со здоровыми структурами.
Лечение
Этиопатогенетического лечения гемимелической эпифизарной дисплазии не существует, поэтому проводимая терапия всегда лишь симптоматическая.
Неизбежному развитию тяжелого артроза и вторичных деформаций у этих больных можно помешать только путем раннего удаления патологических разрастаний хирургическими методами с обязательным и быстрым началом восстановительного лечения.
На выбор тактики хирургического лечения оказывает влияние клиническая картина заболевания, т.е. локализация и степень патологических разрастаний пораженного отдела эпифиза, приводящих к деформации зоны роста и кости в целом, развитию деформирующего артроза и нарушению биомеханики конечности.
Показаниями к операции у детей с гемимелической эпифизарной дисплазией служат боли в пораженном суставе, блокада сустава, нарушение функций конечности, неврологическая симптоматика, вторичные деформации.
Патологические костно-хрящевые разрастания устраняют путем экономных моделирующих резекций пораженных эпифизов с удалением свободно лежащих внутрисуставных тел. Это позволяет восстановить правильное взаимоотношение нагружаемых суставных поверхностей и улучшает функции пораженного сустава.
При вторичных деформациях конечностей (более 7°) выполняют корригирующие остеотомии на межвертельном, надмыщелковом или надлодыжечном уровнях с последующей гипсовой иммобилизацией до полной консолидации отломков. При обширном поражении суставных поверхностей с резко выраженным деформирующим артрозом производят артродез пораженного сустава или эндопротезирование.
Комплекс реабилитационных мероприятий зависит от выраженности патологического процесса и вида хирургического вмешательства. Основной принцип реабилитации при гемимелической эпифизарной дисплазии, как и в целом при операциях на суставах, заключается в раннем начале восстановительного лечения с целью профилактики прогрессирования артроза и вторичных деформаций скелета.
Источники (ссылки)[править]
Ортопедия [Электронный ресурс] : национальное руководство / Под ред. С.П. Миронова, Г.П. Котельникова — 2-е изд., перераб. и доп. — М. : ГЭОТАР-Медиа, 2013. — https://www.rosmedlib.ru/book/ISBN9785970424483.html
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник
Наш генетический код настолько сложный, что практически любая серьезная поломка способна вызвать цепную реакцию и отразиться на человеке не с самой лучшей стороны. Ученые постоянно находят новые заболевания, но, по их собственным словам, на девяносто процентов геном остается неисследованным.
Описание
Синдром Ларсена (МКБ 10 – код М89) – это редко встречающееся генетическое заболевание, которое имеет широкий круг фенотипических проявлений. Наиболее характерными признаками считаются вывихи и подвывихи крупных суставов, наличие пороков развития костей лицевого черепа и проблемы с функциями конечностей. К второстепенным проявлениям относят сколиоз, косолапость, низкий рост и трудности с дыханием.
Синдром Ларсена вызывается точечными мутациями, которые могут произойти как спонтанно, так и быть переданными по наследству по аутосомно-доминантному типу. С изменениями в гене FLNB связывают целую группу заболеваний, проявляющихся нарушениями в скелетной системе. Конкретные проявления могут быть разными даже среди родственников.
Причины
Что же должно произойти в формирующемся организме, чтобы возник синдром Ларсена? Причины этого заболевания все еще скрываются в научном полумраке. Известно только, что для него характерным является аутосомно-доминантное наследование. То есть всего одной копии измененного гена будет достаточно, чтобы передать патологию своим детям, а может быть, даже внукам. Ген может быть получен от родителей (от обоих или от одного) либо являться результатом спонтанной мутации. Риск унаследовать данное заболевание — 50/50, независимо от пола ребенка и количества беременностей.
Измененный ген расположен в коротком плече третьей хромосомы. При желании исследователи могут точно указать место, где наследственная информация подверглась изменениям. В норме этот ген кодирует белок, известный в научных кругах как филамин В. Он играет значительную роль в развитии цитоскелета. Мутации приводят к тому, что белок перестает выполнять возложенные на него функции, и от этого страдают клетки организма.
У людей с данным синдромом возможен мозаицизм. То есть тяжесть и количество проявлений заболевания напрямую зависят от того, сколько клеток оказалось поражено. Некоторые люди могут даже не подозревать о том, что имеют дефект этого гена.
Эпидемиология

Синдром Ларсена с одинаковой частотой развивается как у мужчин, так и у женщин. По весьма приблизительным оценкам, данное заболевание встречается у одного новорожденного из ста тысяч. Это, по счастью, большая редкость. Оценки считаются недостоверными, потому что существуют определенные трудности в выявлении этого синдрома.
Впервые в медицинской литературе заболевание было описано в середине двадцатого века. Лорен Ларсен и соавторы нашли и зафиксировали шесть случаев проявления синдрома у детей.
Симптомы
Синдром Ларсена, как уже упоминалось выше, может по-разному проявляться даже между близкими родственниками. Наиболее характерными признаками заболевания считаются изменения костей лица. К ним относятся: широкая низкая переносица и широкий лоб, плоское лицо, наличие незаращения верхней губы или твердого неба. Кроме того, у детей имеются дислокации крупных суставов (бедренного, коленного, локтевого) и подвывихи плеча.
Пальцы у таких людей короткие, широкие, со слабыми разболтанными суставами. В запястьях могут присутствовать дополнительные кости, которые с возрастом сливаются и нарушают биомеханику движений. У некоторых людей наблюдается такой редкий феномен, как трахеомаляция (или размягчение хрящей трахеи).
Диагностика
Диагноз «синдром Ларсена» ставится только после полного обследования больного, тщательного изучения его истории болезни и наличия характерных рентгенологических симптомов. Кроме того, полноценная радиографическая экспертиза может выявить и сопутствующие аномалии развития скелета, которые имеют косвенное отношение к заболеванию.
Ультразвуковая диагностика еще во внутриутробном периоде может выявить синдром Ларсена. Фото костных образований для хорошо подготовленного специалиста УЗИ может стать отправной точкой для поиска генетических аномалий плода. Так как с первого взгляда нельзя сказать, какая именно болезнь привела к патологии лицевого черепа и костей конечностей, будущей матери рекомендуют сделать амниоцентез и провести генетическую экспертизу для поиска мутации в третьей хромосоме.
Если заболевание подтверждается, но супруги приняли решение о сохранении беременности, то будущей матери рекомендуется провести процедуру кесарева сечения, чтобы не повредить кости ребенка в процессе прохождения его через таз женщины при естественных родах.
Лечение

Лечебные мероприятия направлены не на устранение заболевания, а на снижение клинических проявлений. Этим занимаются педиатры, ортопеды, специалисты по челюстно-лицевой хирургии и генетики. После оценки первоначального состояния ребенка и оценки всех рисков они могут приступить к коррекции имеющихся нарушений.
Самый щадящий вариант, которым лечится синдром Ларсена, – массаж. Он необходим для укрепления мышц и связок, удерживающих суставы, а также для улучшения поддержания спины и выпрямления позвоночника. Но до того как приступить к терапевтическим методам, потребуется ряд операций. Они необходимы для коррекции грубых деформаций скелета или уродств, стабилизации позвонков. При трахеомаляции требуется интубация, а затем и постановка дыхательной трубки (на постоянной основе), которая будет поддерживать проходимость верхних дыхательных путей.
Лечение этого заболевания – процесс длительный, который может растянуться на годы. С взрослением ребенка нагрузка на кости увеличивается, и ему снова может потребоваться физическая реабилитация, лечебная физкультура, а может быть, даже и хирургическое вмешательство.
Источник