Синдром ламберта итона мкб 10

Синдром Итона-Ламберта (Миастенический синдром Ламберта-Итона, МСЛИ, LEMS) — это редкое аутоиммунное заболевание, связанное с нарушением нервно-мышечной передачи, вследствие выработки антител к потенциально- зависимым кальциевым каналам[3]. Заболевание характеризуется слабостью ягодичных и бедренных мышц, птозом, дизартрией, нарушением зрения и периферическими парестезиями. Около 60 % пациентов с LEMS имеют злокачественное онкологическое заболевание (в частности мелкоклеточный рак легкого), поэтому и рассматривается в основном как паранеопластический синдром.
Данное заболевание подробно было изучено в 1965 году Эдвардом Ламбертом и Ли Итоном.
Эпидемиология[править | править код]
Данное заболевание встречается лишь у 3-4 человек на 1 миллион населения. В 67% случаев заболевание представлено опухолевой формой, однако, встречается и аутоимунная форма МСЛИ, которая часто ассоциируется с другими аутоимунными заболеваниями (сахарный диабет, ревматоидный артрит, системная красная волчанка и др.)[4].
Патогенез[править | править код]
В основе патогенеза заболевания лежит выработка аутоантител, триггером которых чаще является опухолевый процесс в организме. Доказано, что мелкоклеточный рак легкого имеет антигены схожие по структуре с холинергическими терминалями нейронов. Вследствие перекрестных иммунологических реакций вырабатывается широкий спектр антител к потенциально зависимым кальциевым каналам, локализованных в пресинаптической мембране синапса. Аутоантитела нарушают механизм высвобождения ацетилхолина в синаптическую щель, что приводит к блокаде проведения импульса.
В литературе отмечены случаи выработки антител к белку синаптотагмин-1, отвечающий за экзоцитоз кальциевых рецепторов, и пресинаптическим мускариновым (M1) ацетилхолиновым рецепторам, усиливающим холинергическую передачу[3].
Клиническая картина[править | править код]
Заболеванием чаще страдают мужчины старше 45 лет. Основными симптомами заболевания являются слабость и повышенная утомляемость мышц проксимальных отделов конечностей и туловища, которые приводят к своеобразной «утиной походке». Позднее может присоединится слабость в дистальных отделах конечностей, а также в стопах. Типичным симптомами для данного заболевания являются сенсорная полиневропатия и прогрессирующая вегетативная дисфункция. Проявления сенсорной полиневропатии следующие: боли в ногах, гипотрофия мышц конечностей, снижение рефлексов, парестезии, гипестезии. К вегетативным нарушениям относятся снижение саливации и потоотделения, которые приводят к сухости слизистых оболочек рта, глаз и кожи, ослабление эректильной функции.
Диагностика[править | править код]
Диагностика заболевания включает в себя следующие этапы:
- Анализ клинической картины;
- Физикальный осмотр;
- Обследовании на исключении опухолевого процесса любой локализации;
- Серологический анализ крови на наличие антител к потенциально зависимым кальциевым каналам;
- Электромиография.
Дифференциальная диагностика[править | править код]
Дифференциальную диагностику проводят со следующими заболеваниями:
- Миастения Гравис;
- болезнь Шегрена;
- полимиозит;
- миопатии эндокринного или метаболического генеза.
Лечение[править | править код]
Наиболее успешным лечением данного заболевания является раннее начало лечения онкопатологии. Назначают химиотерапию и иммуносупрессоры (метотрексат). После исключения ракового процесса назначают симптоматическое лечение. Наиболее эффективным средством лечения холинергических нарушений является гуанидин, однако, данный препарат имеет множество побочных эффектов[3].
- ↑ Disease Ontology release 2019-08-22 — 2019-08-22 — 2019.
- ↑ Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- ↑ 1 2 3 В.В. Пономарев. Аутоимунные заболевания в неврологии. — 1. — Беларуская навука, 2010. — С. 205-209. — 259 с. — ISBN 978-985-08-1134-9.
- ↑ МИАСТЕНИЧЕСКИЙ СИНДРОМ ЛАМБЕРТА-ИТОНА: РЕДКОЕ АУТОИММУННОЕ ЗАБОЛЕВАНИЕ, АССОЦИИРУЮЩЕЕСЯ С РАКОМ | sibac.info. sibac.info. Дата обращения 26 августа 2019.
Источник
Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Лечение
Названия
Название: Синдром Ламберта-Итона.
Синдром Ламберта-Итона
Описание
Синдром Ламберта. Итона — аутоиммунное заболевание, обусловленное поражением пресинаптической мембраны двигательных нервных окончаний и ассоциированное со злокачественными неоплазиями и аутоиммунной патологией. Главным проявлением синдрома является чрезмерная утомляемость и понижение силы мышц, выраженное преимущественно в верхних отделах ног. Диагноз базируется на неврологическом обследовании и данных электронейромиографии. Лечение состоит в удалении опухоли (при ее обнаружении), проведении иммунносупрессивной терапии и сеансов плазмафереза, назначении фармпрепаратов, облегчающих прохождение нервных импульсов по нервно-мышечному синапсу.
Дополнительные факты
Синдром Ламберта-Итона был детально изучен американскими исследователями Ламбертом и Итоном, в честь которых он получил эпонимическое название. Заболевание представляет собой миастенический синдром, ассоциированный с неопластическими и аутоиммунными процессами в организме. Возраст заболевших варьирует в пределах 20-70 лет, но наиболее часто поражаются лица старше 40-летнего возраста. Вначале синдром Ламберта-Итона диагностировался преимущественно у мужчин, и гендерное соотношение составляло 5 случаев заболевания у мужчин к 1 случаю у женщин. Однако современные наблюдения специалистов в области неврологии показали уменьшение этой разницы.
По различным данным, синдром Ламберта-Итона выступает как паранеопластический синдром у 50-75% заболевших; наиболее часто у мужчин (примерно в 70%) и достаточно редко у женщин (до 20%). 80% всех неоплазий, диагностируемых при данном синдроме, составляет мелкоклеточный рак легкого. Причем симптоматика миастенического синдрома может на несколько лет опережать выявление опухолевого процесса. В ряде случаев заболевание Ламберта-Итона сочетается с другими синдромами паранеопластического характера, например, с паранеопластической полиневропатией.
Синдром Ламберта-Итона
Причины
Синдром развивается на фоне злокачественных неоплазий (рак бронха, рак желудка, рак яичников, ретикулосаркома, колоректальный рак, рак простаты и тд ) и аутоиммунных процессов (ревматоидный артрит, болезнь Шегрена, СКВ, аутоиммунный тиреоидит и пр. ). Патогенетическим субстратом заболевания являются аутоиммунные механизмы. У 90% пациентов были выявлены антитела к кальциевым каналам, входящим в структуру, как опухолевых клеток, так и окончаний двигательных нервных волокон. Предположительно мишенью аутоиммунной атаки является пресинаптическая мембрана нервно-мышечного синапса. Ее поражение приводит к уменьшению высвобождения ацетилхолина — медиатора нервно-мышечной передачи. Результатом является нарушение прохождения возбуждения от нервного волокна к мышечной ткани, что клинически проявляется утомляемостью и слабостью мышц.
Симптомы
Основу клинической картины составляет повышенная утомляемость и слабость скелетных мышц, преобладающая в мышцах верхней части ног (мышцы бедра и тазового пояса). Пациенты предъявляют жалобы на слабость в ногах и шаткость, особенно заметные при подъеме по лестнице и продолжительной ходьбе. Могут отмечаться дискомфортные ощущения в области шеи и спины, миалгии, парестезии в дистальных отделах конечностей, вегетативные расстройства (сухость во рту, уменьшение слезопродукции, дистальный гипергидроз, ортостатическая артериальная гипотония). Типична паретичная «утиная» походка. Наблюдается снижение сухожильных рефлексов.
Понос (диарея). Слабость мышц (парез). Сухость во рту.
Диагностика
Диагноз устанавливается неврологом на основании жалоб, неврологического обследования и результатов электронейромиографии. В неврологическом статусе выявляется тетрапарез с акцентом в проксимальных отделах ног, гипорефлексия, легкая дисметрия при выполнении координаторных проб, незначительное снижение глоточного и небного рефлексов.
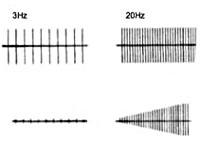
Электронейромиография определяет снижение амплитуды потенциалов действия покоя и временное нарастание амплитуды М-ответа при произвольных мышечных сокращениях или на фоне ритмичной электростимуляции нерва частотой свыше 10 Гц (т. Н. «феномен врабатывания»). При стимуляции частотой 2-3 Гц наблюдается снижение высоты М-ответа, типичное для миастении. Электрофизиологическое исследование позволяет дифференцировать синдром Ламберта-Итона от миастении, миопатии, БАС, полимиозита и другой нервно-мышечной патологии.
Поскольку высока вероятность, что заболевание развилось как паранеопластический синдром, рекомендовано широкое обследование пациента на предмет выявления неоплазии. Проводится анализ крови на онкомаркеры, КТ органов грудной клетки и средостения, МСКТ органов брюшной полости, МРТ головного мозга, КТ или МРТ позвоночника, УЗИ щитовидной железы При отсутствии результатов (невыявлении неоплазии) показаны повторные обследования с интервалом в 6 мес. С целью подтверждения аутоиммунного характера патологии назначаются иммунологические исследования.
Лечение
При паранеопластическом генезе синдрома лечение базируется на ликвидации опухолевого процесса. Если лечение опухоли проходит успешно, то обычно наблюдается регресс симптоматики. При аутоиммунном генезе заболевания проводится симптоматическая терапия фармпрепаратами, угнетающе воздействующими на иммунную систему. В основном используются глюкокортикостероиды. Хороший эффект оказывает плазмаферез, позволяющий отфильтровать из крови циркулирующие в ней аутоантитела.
С целью облегчения нервно-мышечного проведения в схему лечения включают ингибиторы ацетилхолинэстеразы — фармпрепараты, облегчающие нервно-мышечную передачу за счет накопления в синапсе ацетилхолина. К таким препаратам относятся пиридостигмин, ипидакрин. В ряде случаев отмечалось улучшение при применении гуанидина, облегчающего высвобождение ацетилхолина нервными окончаниями. Однако гуанидин не нашел широкого использования вследствие высокой токсичности с побочным воздействием на почки и костный мозг. Менее токсичным фармпрепаратом с аналогичным эффектом является 3,4-диаминопиридин. Его прием может сопровождаться возникновением парестезий, диареи, тахикардии, повышенной бронхиальной секреции. Хотя эффективность 3,4-диаминопиридина доказана клинически, в настоящее время он используется лишь в специализированных медицинских центрах.
Источник
Миастенический синдром Ламберта-Итона (МСЛИ) — редкое аутоиммунное синаптическое нервно-мышечное заболевание, основным симптомом которого является слабость [преимущественно] проксимальных мышц конечностей, обусловленная выработкой антител (АТ [Ig класса G]) к потенциалзависимым кальциевым каналам (ПЗКК [anti-VGCC — voltage-gated calcium channels]) (P/Q- и N-типов) терминалей аксонов (ряд исследователей обнаруживали и антитела к N- и L-типам каналов). АТ ингибируют поступление кальция в терминаль, вызывая нарушение квантового выделения ацетилхолина и блокируя синаптическую передачу.
Обратите внимание! В основе МСЛИ и миастении лежат два противопоставленных друг другу в синапсе дефекта нервно-мышечной передачи, пре- и постсинаптический соответственно, которые объединяет аутоиммунная природа патологических процессов. Патогенез МСЛИ, как было указано выше, обусловлен нарушением квантового выделения ацетилхолина из пресинаптической мембраны аксона. При миастении основной патогенетической мишенью для АТ являются никотиновые ацетилхолиновые рецепторы (АХР) постсинаптической мембраны поперечно-полосатой мышцы (дефект рецепции ацетилхолина).
Большинство исследователей не выявляют у больных с МСЛИ типичных для миастении ауто-АТ к АХР. Вместе с тем в литературе описана группа больных, имеющих комбинацию миастении и МСЛИ (англ.: «overlap myasthenic syndrome»), у которых в различные периоды течения болезни могут преобладать клинические признаки либо миастении, либо МСЛИ и соответственно выявляться АТ и к АХР, и к ПКК.
читайте также пост: Миастения (на laesus-de-liro.livejournal.com) [читать]
По оценке ряда исследователей, заболевание (МСЛИ) наблюдается в 1 — 9 случаях на 1 миллион населения (по данным www.orpha.net распространенность оценивается как 1/250 000- 1/333 300 по всему миру). Небольшая распространенность МСЛИ в общей популяции связана с его поздней диагностикой, так как больные длительное время наблюдаются под «масками» полиневропатии, миопатии, миастении, БАС, а также с низкой выживаемостью пациентов с паранеопластическим типом МСЛИ.
Выделяют [1] паранеопластическую и [2] непаранеопластическую формы МСЛИ (п-МСЛИ и неп-МСЛИ соответственно). п-МСЛИ встречается приблизительно у половины пациентов и связан в основном с мелкоклеточной карциномой (МКК) легкого. Распространенность МСЛИ у пациентов с МКК легкого оценивается на уровне около 3%. Почти у 90% пациентов с п-МСЛИ опухоль выявляется в течение 2 лет после диагностики миастенического синдрома. При этом при центральном мелкоклеточном раке легкого течение болезни бессимптомное в каждом четвертом случае.
МСЛИ, описанный в начале 60-х годов прошлого века как миастенический синдром, иногда сочетающийся, как было указано выше, с МКК легких у злоупотребляющих курением табака пожилых мужчин, по мере накопления клинических наблюдений изменялся с появлением большего числа пациентов без признаков паранеопластического процесса, женщин, а также ранних и даже врожденных форм болезни (сегодня не является столь большой редкостью дебют МСЛИ в детородном возрасте у женщин; сообщения о начале МСЛИ в молодом и детском возрасте встречаются в литературе все чаще).
В связи с этим выделяют 2 пика заболеваемости при МСЛИ: [1] моложе 30 — 40 и [2] старше 50 лет (что отчасти напоминает паттерн миастении, но имеет принципиально иное патогенетическое значение). Патогенез МСЛИ с началом заболевания до 30 — 40 лет, как правило, не связан с неопластической природой, в то время как дебют после 50 лет ассоциируется с МКК легких (80 — 90 % случаев) и в редких случаях с другими неоплазиями (например, опухоль почек, острый лейкоз, ретикулосаркома, злокачественная тимома).
читайте также пост: Паранеопластический неврологический синдром (на laesus-de-liro.livejournal.com) [читать]
Обратите внимание! Несмотря на принятое разделение больных с МСЛИ на 2 группы — п-МСЛИ и неп-МСЛИ, оба эти состояния имеют единую аутоиммунную природу. АТ к ПЗКК P/Q- и N-типов выявляются у 75 — 100 % больных с МСЛИ, сочетающимся с МКК легких, и у 50 — 90 % — у пациентов без признаков неопластического поражения (обратите внимание: возможны серонегативные формы п-МСЛИ и неп-МСЛИ). У больных с МСЛИ как с признаками паранеопластического процесса, так и без такового, помимо специфических аутоантител, выявляются АТ, направленные как против различных антигенных мишеней нервно-мышечного соединения, так и других, например слизистой оболочки желудка, ткани щитовидной железы, клеткам Пуркинье и другим нейрональным структурам, например к декарбоксилазе глутаминовой кислоты). Около 27% пациентов с МСЛИ имеют сопутствующие аутоиммунные заболевания: миастению, в том числе миастению с АТ к специфической мышечной тирозинкиназе, нейромиотонию, тиреоидиты, системную красную волчанку, пернициозную анемию, неспецифический язвенный колит, витилиго, сахарный диабет, ревматоидный артрит. Все это указывает на патогенетическую связь аутоиммунных и паранеопластических процессов при МСЛИ.
МСЛИ клинически характеризуется мышечной слабостью (преимущественно в проксимальных отделах конечностей), гипо- или арефлексией и нарушениями со стороны автономной нервной системы. Обратите внимание: клинически МСЛИ соответствует в большей степени миопатическому синдрому (пациенты с трудом встают со стула [или с корточек] без помощи рук — используют «миопатические приемы», «утиная» походка и др.). В большинстве случаев случаев развиваются признаки вегетативной (автономной) дисфункции, такие как «сухой синдром» (сухость во рту, пониженное потоотделение — ангидроз), импотенция, запор, ортостатическая гипотония и др. Реже встречается слабость дыхательных мышц, вплоть до необходимости применения ИВЛ (искусственной вентиляции легких).
Обратите внимание! Детальное изучение частоты отдельных клинических симптомов у большой группы больных с МСЛИ позволило выявить характерный паттерн болезни, позволяющий дифференцировать МСЛИ от миастении и других нервно-мышечных болезней. У больных с МСЛИ крайне редко выявляется поражение глазодвигательной (птоз, диплопия) и бульбарной мускулатуры (дизартрия, дисфагия, дисфония). Несмотря на жалобы на слабость, объективное снижение силы может быть очень незначительным, а при повторных движениях сила и сухожильные рефлексы увеличиваются [повышаются] ( = синдром «врабатывания») в противоположность тому, что наблюдается у больных миастенией.
Диагноз МСЛИ основывается на клинических и электрофизиологических данных, подтверждается наличием АТ к ПЗКК (в случае «серопозитивности» пациентов радиоиммунологическое исследование выявляет повышение в сыворотке крови концентрации АТ к ПЗКК, в частности, P/Q-типа [норма < 40 пмоль/л]). Как было указано выше, клинический паттерн включает триаду: [1] проксимальную слабость конечностей, [2] а[гипо]рефлексию и [3] автономную дисфункцию (однако следует помнить, что эти три симптома присутствуют не всегда). Классическую электрофизиологическую триаду составляют: [1] уменьшение амплитуды М-ответа; [2] декремент М-ответа при низкой частоте стимуляции (3 имп/с); [3] инкремент М-ответа при высокой частоте стимуляции (40 — 50 имп/с) или после 10-секундного максимального мышечного усилия (на электромиографии [ЭМГ] амплитуда потенциалов действия двигательных единиц в покое ниже нормы, но в отличие от миастении при ритмической электростимуляции двигательного нерва или произвольных сокращениях она возрастает — феномен «врабатывания»). При электронной микроскопии в области нервно-мышечного синапса обнаруживается снижение числа активных зон в области пресинаптических окончаний. Специфических лабораторных изменений нет.
Обратите внимание! Анализ клинических и параклинических данных, выявленных у пациентов с МСЛИ не выявил достоверных клинических, электрофизиологических и иммунологических критериев, позволяющих дифференцировать пациентов с наличием и отсутствием паранеопластического процесса. Изучение концентрации аутоантител к ПКК типа P/Q также не выявило корреляции между уровнем АТ и тяжестью клинических проявлений МСЛИ.
Лечение пациентов с МСЛИ направлено на различные звенья патогенеза заболевания: [1] противоопухолевая терапия (только при доказанном п-МСЛИ); [2] иммуносупрессивная терапия (глюкокортикоиды, внутривенный иммуноглобулин, плазмообмен, цитостатические иммуносупрессанты) для подавления аутоиммунного процесса; [3] симптоматическое лечение (3,4-диаминопиридины), улучшающее нервно-мышечную передачу. Выживаемость больных при сочетании МСЛИ с опухолевым заболеванием в большой степени зависит от своевременности диагностики МСЛИ. Период между временем возникновения первых клинических проявлений МСЛИ и обнаружением опухоли составляет 2 — 5 лет. При этом в большинстве случаев МСЛИ опережает МКК легких; у, примерно, 1/4 пациентов эти заболевания выявляются одновременно; в небольшом числе случаев (примерно 5%) диагностирование МКК легких предшествует развитию МСЛИ.
Подробнее о МСЛИ в следующих источниках:
статья «Миастенический синдром Ламберта-Итона» на www.orpha.net (номер статьи: ORPHA 43393) [читать];
статья «Случай миастенического синдрома Ламберта-Итона на фоне бессимптомной мелкоклеточной карциномы легких» Щербакова Н.И., Пикин О.В., Рудниченко В.А., Галкина О.И., Гуркина Г.Т., Павлов Э.В., Шабалина А.А., Костырева М.В., Глушко В.А., Колбанов К.И., Рудаков Р.В.; ФГБУ «Научный центр неврологии» РАМН, г. Москва; ФГБУ Московский научно-исследовательский онкологический институт им. П.А. Герцена Минздрава РФ (Неврологический журнал, №2, 2014) [читать];
статья «Серонегативный непаранеопластический миастенический синдром Ламберта-Итона» А.Г. Санадзе, Д.В. Сиднев, Д.А. Тумуров; ФГБОУ ВО «Российский национальный исследовательский медицинский университет им. Н.И. Пирогова», Москва; ГБУЗ Москвы «Городская клиническая больница №51» Департамента здравоохранения Москвы, Московский миастенический центр; ГБУЗ Москвы «Научно-практический психоневрологический центр им. З.П. Соловьева» Департамента здравоохранения Москвы (Журнал неврологии и психиатрии, №5, 2017) [читать];
статья «Миастенический синдром Ламберта-Итона на фоне беременности с развитием транзиторного миастенического синдрома новорожденного» Н.И. Щербакова, Г.Т. Гуркина, Л.Ф. Касаткина, В.А. Рудниченко, О.И. Галкина, В.В. Шведков, И.Г. Ретинская, Э.В. Павлов, А.А. Шабалина, М.В. Костырева; ФГБУ «Научный центр неврологии» РАМН, Москва (журнал «Нервно-мышечные болезни» №4, 2013) [читать];
статья «Электронно-микроскопическое исследование скелетных мышц при миастеническом синдроме Ламберта-Итона» Л.Л. Бабакова, О.М. Поздняков; ФГБУ «НИИ общей патологии и патофизиологии» РАМН, Москва (журнал «Нервно-мышечные болезни) №4, 2013) [читать];
статья «Клинический пример паранеопластического синдрома Ламберта-Итона» Лобойко О.И., Консультативная поликлиника ООО «Медицина», г. Харьков (Международный неврологический журнал №3, 2010) [читать]
Источник