Синдром корнелии де ланге клиника

Синдром Корнелии де Ланге (синдром Брахмана-Ланге) — наследственное заболевание, проявляющееся умственной отсталостью и множественными аномалиями развития. Частота заболевания — примерно 1 на 10000[1].
Впервые случай такого заболевания описан в 1916 году немецким врачом В. Брахманом (W. R. C. Brachmann)[2], название синдром получил по имени голландского педиатра Корнелии де Ланге (Cornelia de Lange), в 1933 году описавшей синдром на основе анализа пяти случаев заболевания[3].
Генетика[править | править код]
Синдром Корнелии де Ланге относится к доминантно-наследуемым заболеваниям, большинство случаев являются спорадическими, возникшими de novo. Синдром является генетически гетерогенным. Примерно половина случаев обусловлена мутациями в гене NIPBL, около 5 % случаев — мутациями в гене SMC1A, кодирующем субъединицу белкового комплекса когезина. Один описанный случай связан с мутацией в гене SMC3, который также кодирует одну из субъединиц когезина[4].
В ряде случаев при цитогенетическом исследовании находят микродупликацию локусов q25 — q29 хромосомы 3[5].
Клиническая картина[править | править код]
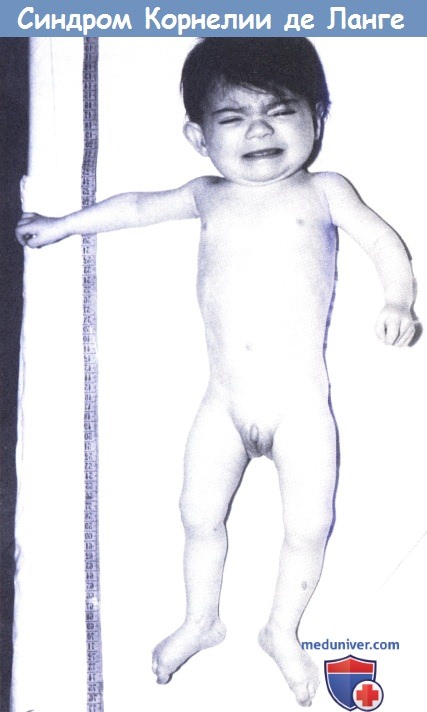
- Микроцефалия (уменьшение размеров черепа более чем на 10 % возрастной нормы);
- Брахицефалия (укорочение черепа в сагиттальном направлении, в результате чего поперечный размер головы увеличивается, а продольный уменьшается);
- Тонкие сросшиеся брови; длинные загнутые ресницы; деформированные ушные раковины; маленький нос, открытые вперед ноздри, атрезия хоан; тонкая верхняя губа;
- Микрогения; высокое нёбо или расщелина нёба; нарушение прорезывания зубов;
- Миопия, косоглазие, астигматизм, атрофия зрительных нервов, колобома зрительного нерва;
- Маленькие кисти и стопы, отсутствие или значительное недоразвитие проксимальных отделов конечностей, вследствие чего кисти и стопы кажутся прикрепленными непосредственно к туловищу, уменьшение количества пальцев;
- Мраморная кожа;
- Гипоплазия сосков;
- Гипертрихоз;
- Судороги;
- Врожденные пороки внутренних органов (сердца, почек, пилоростеноз, крипторхизм) и др.[5]
У части больных наблюдается бег по кругу, стереотипные движения руками и самоповреждения[6].
У всех больных отмечаются отставание в росте, в 80 % случаев — тяжёлая умственная отсталость (имбецильность), в оставшихся 20 % менее выраженные интеллектуальные нарушения[6]; типичны рецидивирующие респираторные инфекции. Выделяют два варианта синдрома: первый (классический) с выраженной пренатальной гипоплазией, значительной задержкой физического и интеллектуального развития, грубыми пороками развития; второй — с аналогичными лицевыми и малыми скелетными аномалиями, но пограничной задержкой психомоторного развития и отсутствием грубых пороков развития[5].
Примечания[править | править код]
- ↑ Opitz J. M., Reynolds J. F. The Brachmann‐de Lange syndrome //American journal of medical genetics. — 1985. — Т. 22. — №. 1. — С. 89-102.
- ↑ Brachmann W. Ein Fall von symmetrischer Monodaktylie durch Ulnadefekt, mit symmetrischer Flughautbildung in den Ellenbeugen, sowie anderen Abnormitäten (Zwerghaftigkeit, Halsrippen, Behaarung): Aus d. Inn. Abt. d. Kinderheilanst. in Dresden; Leiter: Brückner : дис. — Krager, 1916.
- ↑ Cornelia de Lange. Sur un type nouveau de degeneration (typus Amstelodamensis). Archives de medecine des enfants, 1933; 36: 713—719
- ↑ Schrier S. A. et al. Causes of death and autopsy findings in a large study cohort of individuals with Cornelia de Lange syndrome and review of the literature //American Journal of Medical Genetics Part A. — 2011. — Т. 155. — №. 12. — С. 3007-3024
- ↑ 1 2 3 Бородулин В.И., Тополянский А.В. и др. Синдромы и симптомы в клинической практике. Эпонимический словарь-справочник. — М: Эксмо, 2009. — 464 с. — ISBN 978-5-699-33854-2.
- ↑ 1 2 Исаев Д. Н. Умственная отсталость у детей и подростков. Руководство. — СПб: Речь, 2003. — С. 58. — 397 с. — ISBN 5-9268-0212-1.
Источник
Синдром Корнелии де Ланге (синдром Брахмана де Ланге, синдром дегенеративного нанизма амстердамского типа) — этиология, клиника, диагностикаСиндром Корнелии де Ланге (синдром Брахмана де Ланге, синдром дегенеративного нанизма амстердамского типа) представляет собой редкое заболевание, впервые описанное Брахманом в 1916 г., но носящее имя датского педиатра де Ланге. Синдром считается очень редким, частота его составляет всего один случай на 100000 новорожденных (Beck, 1976). а) Патогенез. Синдром Корнелии де Ланге сопровождается мутациями гена NIBPL короткого плеча пятой хромосомы (Gillis et al., 2004; Krantz et al., 2004; Tonkin et al., 2004). Недавно было выявлено, что описанные мутации является основным этиологическим фактором данного синдрома и выявляются у 27-56% пациентов (Yan et al., 2006). Тем не менее, среди пациентов с синдромом Корнелии де Ланге выявляется ряд других хромосомных аномалий (Jackson et al., 1993). Проявления, позволяющие выявить наличие данного синдрома, отмечаются при частичной трисомии дистальной части 3-й хромосомы (3q21-3ter) и других перестройках 3-й хромосомы (DeScipio et al., 2005). Синдром дупликации 3q хромосомы (dup (3q)-синдром) только на первый взгляд имитирует синдром де Ланге (Holder et al., 1994). Данные изменения и изменения, связывающие ген МЕСР2 с синдромом Ретта, доказывают, что следует соблюдать осторожность, рассматривая ген NIBPL как единственную причину развития синдрома де Ланге. Синдром де Ланге может наследоваться доминантным путем; в действительности все случаи синдрома представляют собой вновь возникшие мутации. Риск повторного рождения ребенка с данной патологией составляет 2-5%.
б) Клинические проявления. Синдром де Ланге является мультисистемным заболеванием, проявляющимся пре- и постнатальной задержкой роста, замедленным развитием, характерным дисморфизмом лица, мальформациями конечностей и множественными поражениями органов. Фенотип характеризуется низкой массой при рождении, низким ростом, микроцефалией, генерализованным гирсутизмом и специфическим («любопытным») выражением лица со сросшимися бровями, низкой линией роста волос на лбу и шее, длинными ресницами, вдавленной спинкой носа, длинным подносовым желобком и вывернутыми ноздрями. Характерны плоские лопатообразные кисти и короткие конусовидные пальцы рук. Отмечается клинодактилия пятого пальца кисти. Пальцы стоп располагаются проксимально, а возвышение большого пальца выражено незначительно. Могут встречаться более значимые аномалии конечностей, включая гипоплазию лучевой кости или уменьшение количества пальцев, часто одностороннее. Обычно отмечается микрогнатия (Hawley et al., 1985; Opitz, 1985). Зарегистрированы случаи атрофии зрительного нерва, колобомы зрительного нерва, проптоза и атрезии хоан. Часто встречается желудочно-кишечный рефлюкс, затруднения питания, пониженное слезоотделение и другие поражения глаз. Кроме того, данное заболевание проявляется достаточно характерным поведенческим фенотипом, и в большинстве случаев серьезной или полной необучаемостью (Horsier и Oliver, 2006), часто сочетающейся с самодеструктивным поведением и избеганием социальных контактов, нередко достигая степени синдрома аутизма (Gillberg и Coleman, 2000; Arron et al., 2006; Bhuiyan et al., 2006). Тем не менее, зарегистрированы редкие случаи пограничного или минимально нормального уровня интеллекта. По результатам работы двух исследовательских групп были выявлены значимые различия выраженности или пенетрантности некоторых фенотипов при наличии и отсутствии мутаций (Yan et al., 2006; Selicorni et al., 2007). Различные клинические проявления отмечаются и при разном характере мутаций (миссенс-мутациях и «усеченных мутациях»). Среди пациентов с мутациями отмечается тенденция к более выраженным изменениям массы тела, роста и средней окружности головы при рождении, дисморфизму лица и нарушению речи, чем среди пациентов, у которых мутации отсутствуют. в) Исход. В большинстве случаев тяжелые когнитивные и поведенческие нарушения сохраняются в течение всей жизни. Смертность повышена, частично вследствие нарушения функции желудочно-кишечного тракта в виде регургитации-рвоты-аспирации и развивающейся в тяжелых случаях смертельной пневмонии. Зарегистрированы отдельные случаи, когда пациенты доживали до 40 лет. — Также рекомендуем «Синдром Ди Джорджи (вело-кардио-фациальный синдром, синдром делеции 22q11) — этиология, клиника, диагностика» Редактор: Искандер Милевски. Дата публикации: 4.12.2018 Оглавление темы «Наследственные синдромы в неврологии.»:
|
Источник
Синдром Корнелии де Ланге (синдром Брахмана-Ланге) — наследственное заболевание, проявляющееся умственной отсталостью и множественными аномалиями развития. Частота заболевания — примерно 1 на 10000.
Впервые случай такого заболевания описан в 1916 году немецким врачом В. Брахманом (W. R. C. Brachmann), название синдром получил по имени голландского педиатра Корнелии де Ланге (Cornelia de Lange), в 1933 году описавшей синдром на основе анализа пяти случаев заболевания. Часто используют название синдром Брахмана — Ланге.
Кроме того, встречается название амстердамский синдром, так как в этом городе зафиксировано сразу трое детей, у которых обнаружилась такая патология. Все три наименования – это одно и то же заболевание. Оно встречается на всех континентах, у людей всех рас и этнических групп, с одинаковой частотой как у мальчиков, так и у девочек. За почти сто лет, прошедших с момента первого описания, подробно изучено около 400 случаев данной патологии.

Клиническая картина заболевания
- Очень низкое физическое развитие.
- Низкая масса тела.
- Густые сросшиеся брови.
- Деформированные ушные раковины.
- Густые длинные загнутые ресницы.
- Короткий нос с развернутыми вперед ноздрями.
- Вдавленная переносица.
- Большое расстояние между носом и верхней губой.
- Тонкие губы, опущенные углы рта.
- Череп уменьшен, приплюснут.
- Маленькие кисти, укороченный указательный палец.
- Искривленный мизинец.
- Сросшиеся пальцы стоп.
- Возможно уменьшение числа пальцев.
- Деформация позвоночника и грудины.
- Различные пороки развития внутренних органов (часто различные аномалии строения почек).
- Повышенное оволосение, особенно выраженное в области спины и поясницы.
- Патология развития головного мозга.
- Судороги.

Та или иная степень умственной отсталости диагностируется практически у всех детей с этим синдромом, хотя в литературе описаны случаи со слабо выраженным интеллектуальным дефектом. По данным некоторых авторов, больные с амстердамской карликовостью проявляют стремление к аутоагрессии и склонны к стереотипным движениям, например, бегу по кругу, вращению.
Диагностика
Лабораторные исследования не выявляют постоянных или характерных изменений. Газовая электроэнцефалография всегда указывает на присутствие некоторых диффузных аномалий в обоих полушариях мозга. В виде исключения, встречались и пароксизмальные аномалии биоэлектрических мозговых трасс. Патологоанатомическое обследование выявляет, помимо соматических пороков, обнаруживаемых при макроскопическом исследовании и микроскопические изменения в мозговом веществе, состоящие из кортикальной атрофии и запоздания миелинизации нервных волокон.
Лечение
Специфические методики лечения этого состояния отсутствуют. Младенцам в случаях необходимости делают операции с целью устранения пороков развития, не совместимых с жизнью.
В течение дальнейшей жизни назначаются лечебные процедуры – физиотерапевтические, психотерапевтические, массаж, ношение очков и прочее по симптоматике. Медикаментозное лечение – ноотропы, анаболики, витамины, противосудорожные и седативные препараты.
Профилактика
Профилактикой синдрома, факторы возникновения которого точно не установлены заниматься сложно.
Однако, с учетом известных источников генных мутаций, можно рекомендовать в качестве профилактических мер:
- предотвращение зачатия детей от матери и отца – кровных родственников;
- тщательно обследоваться в случае возможности позднего материнства и отцовства;
- беременным женщинам, избегать заражения вирусными инфекциями, особенно в первом триместре, а в случае заражения применять лекарственную терапию только по назначению врача.
Женщины и мужчины, в семейном анамнезе которых присутствует синдром Корнелии де Ланге, обязательно должны посетить медико-генетическую консультацию. Во время беременности женщинам обязательно нужно обследоваться на наличие протеина-А плазмы крови.
Течение и прогноз заболевания
Течение и прогноз, обычно сдержанные. Смерть может наступить рано (часто даже в течение первых недель жизни) вследствие наслоенных инфекций. Иногда эволюция может продлиться дольше, но, все-таки, с тяжелым отдаленным прогнозом.
Загрузка…
Источник
Медицинский эксперт статьи

х
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Это редкая врожденная патология, характеризующаяся тем, что ребенок рождается со сразу заметными множественными отклонениями от нормы. Впоследствии у младенца обнаруживаются еще и признаки умственной отсталости.
Первым сделал описание синдрома как самостоятельного заболевания немецкий врач В. Брахман в начале ХХ века. Несколько позже педиатр из Нидерландов, Корнелия де Ланге (де Ланж), вела двух маленьких пациенток, страдающих этим заболеванием, и на материалах наблюдений подробно описала его. Эта патология может еще называться синдромом Брахмана-де Ланге или дегенеративный нанизм (карликовость) типа «Амстердам», т.к. трое детей с этим диагнозом жили в столице Нидерландов.
[1], [2], [3]
Код по МКБ-10
Q87.1 Синдромы врожденных аномалий, проявляющихся преимущественно карликовостью
Эпидемиология
Эпидемиология синдрома Корнелии де Ланге: встречается редко, новорожденные с такой патологией появляются примерно в одном случае из 10 – 30 тысяч родов, другие источники называют еще более низкие показатели – один случай из 100 тысяч. Всего на данный момент известно более 400 случаев этого заболевания в разных странах, мальчиков и девочек среди них примерно поровну.
[4], [5]
Причины синдром Корнелии де Ланге
Этиология и патогенез этого синдрома пока не установлены и находятся в стадии изучения. Есть предположения, что заболевание наследственное и может быть обусловлено различными генетическими аномалиями, хотя ген, отвечающий за нарушения внутриутробного развития, и тип его передачи пока не определен (выдвинута гипотеза о мутациях в гене BIPBL (HSA 5p13.1), кодирующем делангин).
Мутации в генах, кодирующие два других белка, участвующих в сплоченности сестринских хроматид, SMC1A и SMC3, были зарегистрированы у 5% и 1% пациентов с синдромом Корнелии де Ланге, соответственно.
Анализ выборок данного заболевания позволяет предположить, что наследование мутантного гена в этом случае не характеризуется примитивной его передачей. Вероятно, со временем усовершенствованное цитогенетическое исследование сможет выявить патологию на хромосомном уровне.
Большинство изученных эпизодов синдрома Корнелии де Ланге являются одиночными, и, обычно, изменений в хромосомном наборе больных не было, хотя изредка выявлялись аномалии – чаще встречалась фрагментарная трисомия по длинному плечу хромосомы 3 и хромосомы 1, а также хромосома 9 имела форму кольца.
Известны и случаи заболевания членов одной семьи, при анализе которых и высказывается предположение об аутосомно-рецессивном способе передачи гена, провоцирующего эту патологию.
Тем не менее, в проявлениях синдрома у членов одной семьи не наблюдается полного или частичного недоразвития конечностей, как в одиночных случаях. На основании этого выдвинута гипотеза о различиях в причинах семейных и одиночных случаев синдрома Корнелии де Ланге.
Влияние возраста отца на частоту появления ребенка с этим заболеванием более чем спорно, поэтому до сих пор непонятно, могут ли вызвать данный синдром одиночные аутосомно-доминантные преобразования генотипа.
[6], [7], [8]
Факторы риска
Факторы риска – наличие в семейном анамнезе этого синдрома, т.к. в этом случае (если предположение о рецессивном способе передачи гена верно) возможность появления следующего ребенка с патологией составляет 25%. Степень вероятности повторения ситуации в одиночных эпизодах, при отсутствии хромосомных мутаций у родителей, теоретически равна 2%.
Предполагается, что преобразования хромосом возникают вследствие тяжелых инфекций и интоксикаций, перенесенных будущей матерью в первые три месяца беременности, побочных действий химиотерапевтических лекарственных средств и некоторых физиотерапевтических процедур. Генным мутациям могут способствовать эндокринные заболевания матери, радиация, солидный возраст отца ребенка либо материнский возраст более 35 лет, а также, когда мать и отец – кровные родственники.
[9], [10]
Симптомы синдром Корнелии де Ланге
Для него характерны многочисленные дефекты развития, которые обычно заметны, хотя иногда выявляются только с помощью диагностических процедур.
Основные признаки синдрома Корнелии де Ланге:
- «причудливое лицо» – густой для новорожденного волосяной покров головы, соединенные брови и длинные загнутые реснички, деформация ушей и маленький носик с открытыми спереди ноздрями, промежуток от верхней губы до кончика носа аномально большой, тоненькая красная кайма верхней губы, уголки губ опущены;
- микроцефалия головного мозга;
- брахицефалия – уменьшение высоты черепа с одновременным увеличением его горизонтального размера;
- патологии полости рта и носоглотки – атрезия хоан, аркообразное небо с расщелиной, сбои в процессе прорезывания молочных зубов.
- дисфункции зрения – страбизм, нарушения формы хрусталика, роговицы, глаза, близорукость, атрофия зрительного нерва;
- укороченные конечности, их эктродактилия, олигодактилия и другие аномалии конечностей;
- кожа мраморной окраски;
- аномалии сосков и гениталий;
- гипероволосение тела;
- эпизодическая судорожная готовность, гипотонус, гипертонус мышц;
- карликовость;
- умственная отсталость разной степени – от незначительных отклонений от нормы (редко) до олигофрении и имбецильности в большинстве случаев.

Первые признаки заболевания визуально заметны у новорожденных. Кроме внешних особенностей, обращает на себя внимание маленький вес ребенка при рождении – он составляет 2/3 веса здорового ребенка, родившегося на аналогичном сроке беременности. Новорожденные имеют проблемы с кормлением и дыханием. С раннего возраста страдают частыми инфекционно-воспалительными заболеваниями дыхательных путей из-за специфического строения носоглотки.
При вскрытиях умерших больных обнаруживаются разнообразные дефекты головного мозга (недоразвитие нижней лобной извилины, расширение желудочков, дисплазия и гипоплазия извилин), гистология нередко показывает выраженную поперечную расчерченность нейронов внешнего зернистого слоя коры больших полушарий и расстройство топографии нейронов мозжечка.
Более, чем в половине всех случаев амстердамскому нанизму сопутствуют дефекты в структуре сердца (аортолегочное окно, незарощенная перегородка, разделяющая как предсердия, так и желудочки, часто в комбинации с сосудистыми нарушениями, тетрада Фалло), дефекты в структуре ЖКТ (в основном – нарушения поворота кишечника), мочеполовой системы (кистозные образования почек, одиночные и множественные, иногда – подковообразная почка и гидронефротические ее изменения, крипторхизм, двурогая матка).
Это заболевание, характеризующееся множеством дефектов развития, является по своей сути пока еще не раскрытой генетической аномалией, которая начинается в период формирования эмбриона. Процесс, запущенный патогенным фактором, продолжается и усугубляется в дальнейшем, после рождения ребенка. Стадии заболевания идут рука об руку с биохимическими патологиями в мозговых нейронах на протяжении всех ступеней созревания организма. Подобные поражения сопровождаются умственной отсталостью, а имеющиеся множественные поведенческие и внешние отклонения у больного еще не свидетельствует об окончании процесса во внутриутробном периоде.
[11], [12]
Формы
Современная психиатрия классифицирует следующие виды данного синдрома:
- Классический (первый), когда все симптомы ярко проявляются: специфическая внешность, множественные пороки развития, заметная умственная отсталость.
- Стертый вид (второй), при котором имеют место те же дефекты лица и туловища, однако не совместимых с жизнью аномалий внутренних органов нет, нарушения моторики, психики и интеллекта слабо выражены.
По наблюдениям родителей, дети с таким заболеванием ни в каком возрасте не просятся в туалет, склонны к раздражительности, постоянно совершают бессмысленные, не характерные для здоровых детей поступки: рвут или едят бумагу, разламывают все, что попадается им на глаза, передвигаются кругами. Это им приносит успокоение.
[13], [14], [15], [16]
Осложнения и последствия
Последствия и осложнения наличия синдрома де Ланге неблагоприятны, люди очень зависимы от окружающих, жить самостоятельно, без постоянной помощи они не в состоянии, в классических случаях возможна гибель от какой-либо патологии развития внутренних органов еще во младенчестве.
[17], [18], [19]
Диагностика синдром Корнелии де Ланге
На современном этапе развития диагностики невозможно пока обнаружить наличие данной патологии у эмбриона. Фактором риска развития синдрома является отсутствие в сывороточной крови беременной женщины протеина-А плазмы (РАРР-А), который в норме вырабатывается в период беременности в больших количествах. Однако, точно диагностировать присутствие заболевания у эмбриона только по результатам этого теста невозможно, т.к. в 5 % случаев нормальных беременностей наблюдается ложнопозитивный результат, а хромосомные отклонения у плода обнаруживают только в 2-3 % случаев уменьшения уровня этого белка.
Амстердамскую карликовость определяют у новорожденных по характерным внешним признакам.
Множественные дефекты и аномалии, не совместимые с жизнью, должны быть вовремя диагностированы, чтобы можно было осуществить оперативное вмешательство, необходимое для сохранения жизни.
Инструментальная диагностика проводится с помощью магниторезонансной томографии, ультразвукового и рентгенографического исследования, риноскопии и прочих современных методов диагностики по необходимости.
Больному делают как стандартные клинические анализы, так и цитогенетические.
Диагностика проводится в два этапа: клиническое обследование состояния новорожденного, соответствующее современным методикам, и дифференциальная диагностика конкретной генетической патологии. Она базируется на дифференциации подобных поражений с наиболее типичными при данном синдроме симптоматическими проявлениями.
Диагностика синдрома де Ланге подчас спорна, поскольку попадаются дети с умственной отсталостью и малым количеством дефектов – признаков данного заболевания. Так как бесспорного биологического способа подтверждения диагноза не существует, невозможно точно определить, относятся ли эти эпизоды к данному синдрому.
[20], [21]
Лечение синдром Корнелии де Ланге
Специфические методики лечения этого состояния отсутствуют. Младенцам в случаях необходимости делают операции с целью устранения пороков развития, не совместимых с жизнью.
В течение дальнейшей жизни назначаются лечебные процедуры – физиотерапевтические, психотерапевтические, массаж, ношение очков и прочее по симптоматике. Медикаментозное лечение – ноотропы, анаболики, витамины, противосудорожные и седативные препараты.
Профилактика
Профилактикой синдрома, факторы возникновения которого точно не установлены заниматься сложно.
Однако, с учетом известных источников генных мутаций, можно рекомендовать в качестве профилактических мер:
- предотвращение зачатия детей от матери и отца – кровных родственников;
- тщательно обследоваться в случае возможности позднего материнства и отцовства;
- беременным женщинам, избегать заражения вирусными инфекциями, особенно в первом триместре, а в случае заражения применять лекарственную терапию только по назначению врача.
Женщины и мужчины, в семейном анамнезе которых присутствует синдром Корнелии де Ланге, обязательно должны посетить медико-генетическую консультацию. Во время беременности женщинам обязательно нужно обследоваться на наличие протеина-А плазмы крови.
[22], [23], [24]
Прогноз
Срок жизни, отмеренный людям с этим заболеванием, зависит от многих факторов, главными из них можно назвать – степень тяжести пороков жизненно важных органов, их ранняя диагностика и качество хирургических вмешательств по их ликвидации.
При аномалиях развития, несовместимых с жизнью, ребенок умирает на первой неделе жизни. В случае их незначительности или своевременного устранения хирургическим путем, больной с синдромом Корнелии де Ланге может прожить достаточно долго. Прогнозирование осложняется отсутствием сопротивляемости организма больных с данным синдромом ординарным, неопасным для обычных людей инфекциям, например, вирусным, которые тоже становятся причиной ранней смерти таких больных.
Средняя продолжительность жизни примерно 12-13 лет, по некоторым источникам больные со стертой формой заболевания или удачно проведенными операциями по устранению дефектов развития иногда доживали до пятого-шестого десятка лет.
[25], [26]
Источник