Синдром клайнфельтера по мкб 10

Рубрика МКБ-10: Q98.0
МКБ-10 / Q00-Q99 КЛАСС XVII Врожденные аномалии пороки развития, деформации и хромосомные нарушения / Q90-Q99 Хромосомные аномалии, не классифицированные в других рубриках / Q98 Другие аномалии половых хромосом, мужской фенотип, не классифицированные в других рубриках
Определение и общие сведения[править]
Синдром Клайнфельтера — это клиническое проявление полисомии по X-хромосоме у мужчин. Чаще всего наблюдается кариотип 47,XXY (классический вариант синдрома), но встречаются и более редкие кариотипы: 48,XXXY; 49,XXXXY; 48,XXYY; 49,XXXYY. Наличие в кариотипе не менее двух X-хромосом и одной Y-хромосомы — самая распространенная причина первичного гипогонадизма у мужчин.
Распространенность синдрома Клайнфельтера около 1:500.
Этиология и патогенез[править]
Примерно у 10% больных с синдромом Клайнфельтера наблюдается мозаицизм 46,XY/47,XXY. Поскольку в формировании фенотипа участвует клон клеток с нормальным кариотипом, больные с мозаицизмом 46,XY/47,XXY могут иметь нормально развитые половые железы и быть фертильными. Добавочная X-хромосома в 60% случаев наследуется от матери, особенно при поздней беременности. Риск наследования отцовской X-хромосомы не зависит от возраста отца.
Клинические проявления[править]
Производные вольфова протока формируются нормально. В детском возрасте нарушения развития яичек незаметны и могут не выявляться даже при биопсии. Эти нарушения обнаруживают в пубертатном периоде и позднее. В типичных случаях при биопсии яичка у взрослых находят гиалиноз извитых семенных канальцев, гиперплазию клеток Лейдига, уменьшение численности или отсутствие клеток Сертоли; сперматогенез отсутствует. Больные, как правило, бесплодны (даже если есть признаки сперматогенеза). Формирование вторичных половых признаков обычно нарушено: оволосение лица и подмышечных впадин скудное или отсутствует; наблюдается гинекомастия; отложение жира и рост волос на лобке по женскому типу. Как правило, психическое развитие задерживается, но у взрослых нарушения интеллекта незначительны. Нередко встречаются нарушения поведения, эпилептическая активность на ЭЭГ, эпилептические припадки. Сопутствующие заболевания: рак молочной железы, сахарный диабет, болезни щитовидной железы, ХОЗЛ.
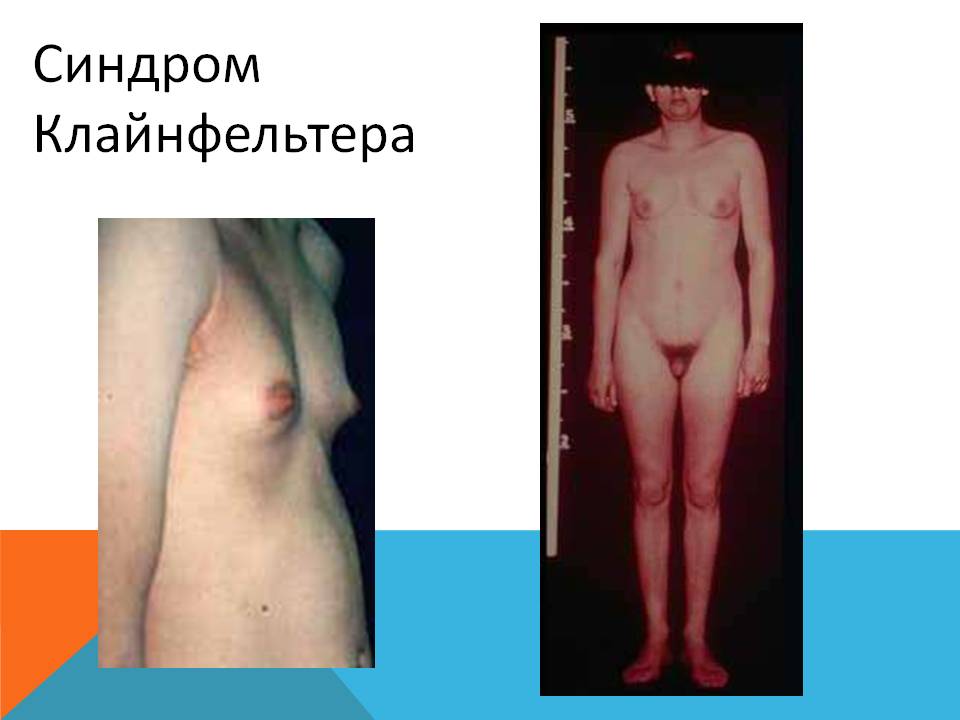
Для синдрома Клайнфельтера характерен фенотипический полиморфизм. Наиболее частые признаки: высокорослость, непропорционально длинные ноги, евнухоидное телосложение, маленькие яички (длинная ось < 2 см).
Синдром Клайнфельтера, кариотип 47,XXY: Диагностика[править]
Дифференциальный диагноз[править]
Синдром Клайнфельтера, кариотип 47,XXY: Лечение[править]
Способы лечения бесплодия при синдроме Клайнфельтера пока не разработаны. Заместительную терапию тестостероном обычно начинают с 11—14 лет; при дефиците андрогенов она существенно ускоряет формирование вторичных половых признаков. У взрослых больных на фоне лечения тестостероном повышается половое влечение. При гинекомастии может потребоваться хирургическое вмешательство. Психотерапия способствует социальной адаптации больных с синдромом Клайнфельтера и больных с другими аномалиями половых хромосом.
Профилактика[править]
Прочее[править]
Источники (ссылки)[править]
Дополнительная литература (рекомендуемая)[править]
1. Buyse ML. Birth Defects Encyclopedia. Cambridge: Blackwell, 1990.
2. Emery A, Rimoin D. Principles and Practice of Medical Genetics (2nd ed). New York: Churchill Livingstone, 1990.
3. Gorlin RJ, et al. Syndromes of the Head and Neck (3rd ed). New York: Oxford University Press, 1990.
4. Hall JG, et al. Handbook of Normal Physical Measurements. New York: Oxford University Press, 1989.
5. Jones KL. Smith’s Recognizable Patterns of Human Malformation (4th ed). In M Markowitz (ed.), Major Problems in Clinical Pediatrics (vol VII). Philadelphia: Saunders, 1988.
6. McKusick V. Mendelian Inheritance in Man (10th ed.). Baltimore: Johns Hopkins University Press, 1992.
7. Rimoin DL. Disorders of the Endocrine Glands. In AA Dietz (ed.), Genetic Disease: Diagnosis and Treatment. Proceedings of the Fifth Arnold O. Beckman Conference in Clinical Chemistry. Monterey, CA: The Association for Clinical Chemistry, 1983.
8. Taybi H, Lachman RS. Radiology of Syndromes, Metabolic Disorders, and Skeletal Dysplasias (3rd ed). Chicago: Yearbook, 1990.
9. Vogel F, Motulsky AG. Human Genetics: Problems and Approaches (2nd ed). New York: Springer, 1986.
10. Wiedmann H-R, et al. Atlas of Clinical Syndromes—A Visual Aid to Diagnosis (2nd ed). St. Louis: Mosby, 1989.
11. Wynne-Davis R, Hall CM, Apley AG. Atlas of Skeletal Dysplasias. Edinburgh: Churchill Livingstone, 1985.
Действующие вещества[править]
Источник
Содержание
- Описание
- Симптомы
- Диагностика
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Синдром Клайнфельтера.
Внешний вид больного с синдромом Клайнфельтера
Описание
Эта хромосомная патология бывает довольно часто: она встречается в среднем 1:850 новорожденных мужского пола и у 1-2,8% больных олигофренией, чаще при неглубоком интеллектуальном снижении. Среди мужчин, страдающих бесплодием бесплодием, более, более 10% имеют дополнительную Х-хромосому. Средней возраст родителей при рождении ребенка с болезнью Клайнфельтера повышен: он равен 35,5 года у отцов.
Симптомы
Внешний вид новорожденных с синдромом обычный. Изменения, как правило, начинают клинически проявляться в препубертатном и пубертатном возрасте.
Взрослые мужчины имеют высокий рост, евнухоидное телосложения (длинные ноги, высокая талия, относительно широкий таз, отложение жира по женскому типу), склонность к ожирению, гинекомастию. Как специфический для заболевания симптом, не встречающийся при других формах гипогонадизма, отличают относительно короткие руки, их размах не более чем на 2-3 см превышает рост, в то время как при иных вариантах недостаточности гормональной активности половых желез это различие составляет не менее 4 Данный симптом выявляется еще в допубертатном периоде. Подмышечное оволосение выражено недостаточно, на лобке оволосение по женскому типу, растительность на лице незначительная или отсутствует. У больных часто встречается различные диспластические признаки: уплощенный затылок, гипертелоризм, эпикант, выступающие надбровные дуги, высокое небо, неправильный рост зубов, приросшие мочки, клинодактилия мизинцев кистей. Мускулатура развита слабо, плечи узкие, грудная клетка уплощена. Половой член нормальных размеров или уменьшен, яички значительно уменьшены, плотность яичек значительно повышена, безболезненные.
Умственная отсталость отмечается у 25-50 % больных. Интеллектуальная недостаточность выражена нерезко, в основном это пограничная умственная отсталость и дебильность различной степени. Больным свойственны астенические проявления и черты психического инфантилизма: неустойчивость внимания, повышенная утомляемость, снижение работоспособности, повышенная внушаемость Для синдрома Клайнфелтера характерна определенная диссоциация между недоразвитием интеллекта и незрелостью эмоционально-волевой сферы.
Лимфоцитоз. Потливость.
Признаки синдрома Клайнфельтера
Диагностика
Среди дерматоглифических признаков нередко встречаются поперечная складка, дистальное расположение трирадиуса, увеличение частоты дуг на пальцах.
Исследование спермы выявляет зрелые формы сперматозоидов только в очень редких случаях. Как правило, обнаруживается олиго- или азооспермия. В пунктате яичка находят гиперплазию клеток Лейдига, гиалинизацию семенных канатиков. Уровень фолликулостимулирующего гормона значительно повышен.
Кариологические находки разнообразны: в большинстве случаевобнаруживается классический кариотип 47, ХХУ; встречаются и кариотипы 48, ХХХУ; 49, ХХХХУ, а также различные формы мозайцизма: 47, ХХУ/46, ХУ; 47, ХХУ/ 46, ХХ; 47, ХХУ/46,ХУ/46,ХХ.
При трех лишних Х-хромосомах (49, ХХХХУ) симптомокомплекс настолько отличается от классического синдрома Клайнфельтера, что некоторые клиницисты выделяют его в отдельный синдром-тетрасомию Х. При этом синдроме отмечается низкая масса тела при рождении (в среднем 2600г). Для внешнего облика характерно овальное лицо, гипертелоризм, косой размер глаз, эпикант, косоглазие, спинка носа несколько уплощена, вдавлена, а кончик носа вздернут. Рот большой, четко очерчен, иногда приближается к треугольной форме. Ушные раковины большие, недоразвитые, расположены ниже обычного. Шея короткая, широкая. Со стороны костно-мышечной системы выявляют затруднение сгибания в локтевых суставах, ограничение супинации, клинодактилия 5 пальцев кисти, нередко искривление шейки бедра с его вальгусным положением, сандалевидная щель. Резко выражен гипогонадизм, при биопсии находят те же изменения, что и у больных с кариотипом 47,ХХУ. Умственная отсталость при тетрасомии ХУ встречается во всех случаях и соответствует глубокой дебильности или имбецильности.
Лечение
Лечение синдрома Клайнфельтера, главным образом гормональное, предпочтительнее начинать с 10-12 лет, терапия препаратами мужских половых гормонов улучшает физическое состояние. Обычно назначают 1% или 5% растворы тестостерона пропионата по 0,5-1 мл внутримышечно 2-3 раза в неделю или сустанон-250 по 0,5-1 мл 1 раз в месяц, также 5-10 мг метилтестостерона сублингвально 2-3 раза в день. Однако при этом необходимо помнить, что гормональное лечение повышает сексуальное влечение. Выраженную гинекомастию лечат хирургическим путем. При неглубоком интеллектуальном снижении применяют психостимуляторы и нейрометаболические препараты. Для стимуляции роста волос на лице используют растирки и мази, содержащие андрогены. Риск повторного рождения ребенка с синдромом Клайнфельтера не превышает 1%, если кариотип родителей нормальный.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Медицинский эксперт статьи

х
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Синдром Клайнфельтера, 47, XXYявляется клиническим примером поражения половых хромосом.
Болезнь Клайнфельтера характеризуются наличием как минимум одной лишней X хромосомой у мальчиков, что приводит к нарушению полового созревания у них. Клинически впервые описан Клайнфельтером в 1942 г. Популяционная частота составляет 1 : 1000 лиц мужского пола. Синдром Клайнфельтера встречается примерно у 1/800 родившихся живыми мальчиков. Лишнюю Х-хромосому ребенок получает от матери в 60 % случаев.
 [1], [2], [3]
[1], [2], [3]
Код по МКБ-10
Q98.0 Синдром Клайнфелтера, кариотип 47,ХХУ
Что вызывает болезнь Клайнфельтера?
В большинстве случаев неправильное расхождение половых хромосом происходит в гаметах родителей. Имеют место и мозаичные варианты, например 47, XXY/46, XY.
Синдром Клайнфельтера обусловлен хромосомной патологией, представленной в наиболее типичном варианте как 47ХХУ. Значительно реже встречаются мозаичные формы — 46ХУ/47ХХУ. Как казуистические варианты кариотипа описаны формы 48ХХХУ, 47ХХУ/46ХХ, 47ХХУ/45ХО. Имеется также наблюдение пациента с кариотипом 47ХХУУ46ХХ/45ХО. Причиной данных хромосомных аномалий — дополнительной Х-хромосомы в мужском кариотипе — может являться нерасхождение Х-хромосомы в период первого или второго мейотического деления или нарушение митотического расхождения хромосом в период развития зиготы (мозаичные варианты). Метод ДНК-анализа позволил выявить, что 53% больных с синдромом Клайнфельтера имели дополнительную хромосому отцовского происхождения, являвшуюся следствием нерасхождения в период первого мейотического деления. 43% больных имели дополнительную хромосому материнского происхождения как результат патологии первого и второго мейотического деления. По-видимому, не имеется различий фенотипа у пациентов, имеющих дополнительную материнскую или отцовскую Х-хромосому Частота рождения мальчиков с синдромом Клайнфельтера увеличивается по мере увеличения возраста матери. Подобной зависимости от возраста отца не выявлено. Наличие в мужском кариотипе дополнительной Х-хромосомы не влияет на дифференцировку тестикул и формирование гениталий по мужскому типу. Однако жизнедеятельность герминативных клеток нарушается, сперматогенез отсутствует. Причиной этого является активность дополнительной Х-хромосомы в герминативных клетках, имеющих в норме гаплоидный набор хромосом. Было показано, что в герминативных клетках яичника плода у девочек перед вступлением в мейоз происходит реактивация второй Х-хромосомы (в норме активирована только одна). У мальчиков с кариотипом ХХУ также сохранен пред мейотический процесс реактивации второй Х-хромосомы, однако процесс расхождения нарушен, и герминативная клетка может содержать две активные Х-хромосомы, что приводит к ее гибели уже в первые дни после реактивации Х-хромосомы. У взрослых мужчин с синдромом Клайнфельтера при анализе клеток спермы единичные сохранные герминативные клетки имели только нормальный гаплоидный хромосомный набор.
Патогенез синдрома Клайнфельтера
Наличие лишней X хромосомы приводит к аплазии эпителия тестикул, которые в дальнейшем гиалинизируются. Это приводит у взрослых больных к азооспермии и бесплодию.
[4], [5], [6], [7]
Симптомы синдрома Клайнфельтера
При рождении синдром Клайнфельтера клинически не проявляется. Клинических вариантов, касающихся как аномалий полового статуса, так и соматических нарушений при синдроме Клайнфельтера, описано достаточно много. Общей закономерности влияния кариотипа на фенотип не выявлено, но пациенты, имеющие мозаичный кариотип с нормальным мужским клоном 47ХХУ/46ХУ, имеют менее тяжелые нарушения.
Первые отчетливые фенотипические признаки заболевания появляются в пре- и пубертатном периодах онтогенеза. До пубертатного возраста у мальчиков могут выявляться крипторхизм (чаще двусторонний) и маленькие размеры полового члена. У 50% мальчиков отмечается умеренная задержка умственного развития, сопровождающаяся нарушениями поведения, трудностями контакта со сверстниками. Мальчики обычно имеют длину тела выше средневозрастных показателей. Характерны относительно длинные конечности, избыточное жироотложение по женскому типу (евнухоидное телосложение).

Поздно появляются вторичные половые признаки. Наиболее характерным симптомом синдрома Клайнфельтера является гипоплазия яичек и полового члена (гипогонадизм и гипогенитализм). У 50% больных в периоде полового созревания выявляется гинекомастия. Имеет место неглубокое снижение интеллекта, что сказывается на школьной успеваемости. Взрослые пациенты склонны к алкоголизму, наркомании, гомосексуализму и асоциальному поведению, особенно в условиях стресса.
Пубертат, как правило, начинается в обычном возрасте, однако часто оволосение на лице низкое. У таких детей наблюдается предрасположенность к нарушениям обучения, у многих отмечается сниженный вербальный интеллект, нарушены слуховое восприятие и обработка информации, а также навыки чтения. Клиническая вариабельность значительная, многие мальчики и мужчины с кариотипом 47, XXY имеют обычную внешность и нормальный интеллект.
В пубертатном возрасте вторичное оволосение появляется в обычные сроки, отмечается также увеличение полового члена. Однако объем тестикул увеличивается незначительно, не превышая, как правило, 8 мл; яички имеют плотную консистенцию. Пубертатная гинекомастия, часто достаточно ранняя, выявляется у 40-50% мальчиков В дальнейшем у этих пациентов повышается риск развития карциномы молочных желез. Костное созревание обычно соответствует возрасту к моменту инициации пубертата, однако позже дифференцировка костей скелета задерживается в связи с недостаточностью секреции тестостерона. Линейный рост конечностей продолжается до 18-20 лет, что приводит к формированию евнухоидных пропорций тела, конечный рост больных, как правило, выше роста родителей. Постпубертатная инволюция тестикул приводит к гипогонадизму и потере фертильности. При гистологическом исследовании выявляется гиалиноз семенных канальцев и отсутствие сперматогенеза. Количество клеток Лейдига может быть нормальным, однако с возрастом они подвергаются атрофии.
Помимо симптомов нарушения полового развития у больных на синдром Клайнфельтера может выявляться целый ряд врожденных аномалий костном ткани: клинодактилия, деформация грудины, cubitus valgus, coxa valga, гиперте лоризм, микрогнатия, «готическое» небо и др. Нередко заболевание сопровождается врожденными пороками сердечно-сосудистой системы. У больных достаточно часто выявляются злокачественные ново образования, в частности имеются сведения о высокой частоте герминативно-клеточных опухолей.
В 15 % случаев наблюдается мозаицизм. Эти мужчины могут иметь детей. У некоторых мужчин может быть 3,4 и даже 5 Х-хромосом вместе с одной Y-хромосомой. С увеличением числа Х-хромосом возрастает тяжесть умственной отсталости и пороков развития.
[8], [9]
Классификация синдрома Клайнфельтера
В диагнозе указывается цитогенетический вариант синдрома.
[10], [11], [12]
Диагностика синдрома Клайнфельтера
Часто синдром Клайнфельтера обнаруживают при обследовании по поводу бесплодия (вероятно, все 47, XXY мужчины стерильны). Развитие яичек варьирует от гиалинизированных нефункционирующих тубулярных структур до некоторой продукции сперматозоидов; часто отмечается повышенная экскреция с мочой фолликулостимулирующего гормона.
При наличии фенотипических признаков синдрома Клайнфельтера определяют половой хроматин. Если проба положительная, то показано проведение кариотипирования. При этом в большинстве случаев выявляют кариотип 47, X X Y либо его мозаичный вариант. Однако встречаются и другие цитогенетические варианты синдрома, например 48, X X X Y ; 48, XXYY.
[13], [14], [15], [16], [17], [18], [19]
Особенности гонадотропной и гонадной функций
В допубертатном возрасте показатели ЛГ, ФСГ и Т у мальчиков с синдромом Клайнфельтера обычно соответствуют норме. К началу пубертата уровень ФСГ повышается и к 14-15 годам уже значительно превышает норму. Уровень тестостерона к моменту пубертата обычно повышается, но его концентрация не достигает нормативных показателей. Уровень ЛГ в период пубертата соответствует норме, но в последующем по мере снижения уровня тестостерона концентрация ЛГ возрастает. Реакция ЛГ и ФСГ на введение ГнРГ обычно носит гиперэргический характер уже на ранних стадиях пубертата
Процесс формирования андрогеновой недостаточности, являющейся вторичной по отношению к первичному повреждения герминативного эпителия тестикул, в настоящее время окончательно не выяснен. Ранняя гибель сперматогенного эпителия приводит к недостаточности клеток сертоли, секретируюших ингибин — естественный регулятор секреции ФСГ у мужчин. В результате уровень ФСГ у больных повышен уже с раннего пубертатного возраста. Однако выработка тестостерона и секреция ЛГ в первые годы пубертата и постпубертатного возраста не нарушены, только в дальнейшем отмечаются снижение секреции тестостерона и повышение секреции ЛГ — развитие гипергонадотропного гипогонадизма. Очевидно, что герминативный эпителий и клетки Сертоли оказывают определенное трофическое воздействие на интерстициальные клетки Лейдига, а и отсутствие их трофического влияния делает невозможным нормальную секрецию тестостерона.
[20], [21], [22], [23], [24], [25], [26], [27], [28], [29]
Дифференциальный диагноз синдрома Клайнфельтера
В случаях наличия признаков синдрома Клайнфельтера при нормальном кариотипе (46, XY) необходимо исключать другие формы гипогонадизма.
[30], [31], [32]
Какие анализы необходимы?
Как лечится болезнь Клайнфельтера?
В периоде полового созревания проводят курсы андрогенов, что способствует формированию вторичных половых признаков, однако бесплодие не излечивается.
Подросткам с синдромом Клайнфельтера, несмотря на парциальный андрогеновый дефицит, терапию препаратами эфиров тестостерона по стандартной схеме следует назначать с 13-14 лет Препараты андрогенов значительно улучшают адаптацию и интеллект подростка, предотвращают развитие евнухоидизма. Длительное наблюдение за подростками с синдромом Клайнфельтера показало, что ранняя терапия препаратами тестостерона значительно повышает интеллект взрослых пациентов, их трудоспособность и социальную адаптацию.
[33]
Оценка эффективности лечения
Критерий эффективности лечения — развитие вторичных половых признаков.
[34], [35], [36], [37], [38]
Осложнения и побочные эффекты лечения
Введение эфиров тестостерона может вызывать задержку жидкости, возбуждение в первые дни после инъекции.
Диспансерное наблюдение осуществляется эндокринологом.
[39], [40], [41], [42], [43], [44]
Какой прогноз имеет синдром Клайнфельтера?
Синдром Клайнфельтера имеет разный прогноз и зависит от формы заболевания, сочетанных гормональных и соматических нарушений. Заместительная терапия половыми гормонами пожизненная.
Источник