Синдром клайнфельтера клиника диагностика лечение


Синдром Клайнфельтера — распространенный генетический недуг, обусловленный дефицитом гормона тестостерона. Клиническую картину и особенности патологии впервые описали в 1942 году американские врачи Г. Клайнфельтер и Ф. Олбрайт. В честь одного из них аномалия получила свое название. Эта хромосомная болезнь регистрируется у одного или двух мальчиков из тысячи новорожденных. У больных в кариотипе присутствует хотя бы одна дополнительная женская хромосома. Патология не создает проблем таким детям до начала пубертатного периода.
Синдром Клайнфельтера — самая распространенная генетическая патология, а также не менее популярное эндокринное заболевание, которое часто остается нераспознанным. В структуре эндокринной патологии оно уступает лишь сахарному диабету и тиреотоксикозу. Первичная форма гипогонадизма сопровождается плохим функционированием секреторной ткани яичек, неполным развитием внутренних и наружных половых органов, недоразвитием вторичных половых признаков, нарушением белкового и жирового обмена веществ. Заболевание не передается по наследству, поскольку больные мужчины абсолютно бесплодны.
Диагностика синдрома Клайнфельтера основывается на данных кариотипирования, результатах определения в крови половых гормонов, спермограммы, УЗИ мошонки и морфологического изучения биоптата яичек. В настоящее время этиологическое лечение хромосомных аномалий не разработано. Современные терапевтические методики позволяют сгладить основные симптомы болезни и нормализовать социальную жизнь носителей более двух половых хромосом. Больным назначают гормоны, проводят оперативное вмешательство — мастэктомию. Синдром Клайнфельтера – неизлечимая патология.

Этиология
Синдром Клайнфельтера обусловлен мутацией генов, приводящей к удвоению женской половой хромосомы в кариотипе мужчины. Нерасхождение половых хромосом в процессе мейоза либо митоза может быть обусловлено различными факторами. Самыми распространенными среди них являются:
- Вирусы,
- Неполноценность иммунной системы матери или отца,
- Плохое экологическое состояние окружающей среды,
- Дети от родственных браков,
- Ранний или поздний возраст матери,
- Наследственные патологии в предыдущих поколениях.

Мальчики с синдром Клайнфельтера вместо нормального мужского генотипа ХУ приобретают одну У хромосому и несколько Х хромосом. Такое изменение генетического набора приводит к появлению особых внешних данных, незначительному снижению интеллекта и развитию целого ряда сопутствующих заболеваний.
Повышение концентрации фолликулостимулирующего и лютеинизирующего гормонов в крови приводит к фиброзу, гиалинозу и атрофии семенных канальцев. Яички перестают развиваться, становятся маленькими и плотными. Облитерация семенных канальцев заканчивается развитием азооспермии и бесплодия.
Клиника

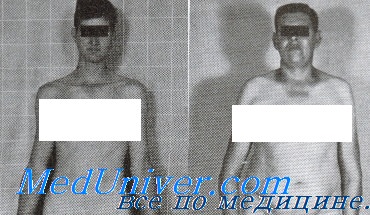
Мальчики с синдромом Клайнфельтера при рождении имеют нормальный рост и вес. Клинические признаки заболевания у них тоже отсутствуют: мошонка и половой член располагаются правильно и симметрично. Обнаружить патологию у новорожденных и грудных детей невозможно. Клиника синдрома Клайнфельтера становится явной только после начала пубертатного периода. Заподозрить заболевание можно и раньше по ряду характерных признаков. У больных мальчиков ноги длиннее, чем у сверстников, талия расположена несколько выше, мускулатура развита слабо. Узкие плечи и широкие бедра придают фигуре определенную женственность. Отложение жира также происходит по женскому типу. Диспропорциональное телосложение — один из характерных признаков данного наследственного заболевания.
Мальчики с синдромом Клайнфельтера подвержены часто возникающим вирусным респираторным заболеваниям. Это первый тревожный признак, заставляющий родителей и педиатров обратить особое внимание на ребенка. Больные дети поздно начинают ползать, сидеть, ходить, говорить.
Мальчики заметно прибавляют в росте между 6—7 годами. Большинство из них имеют атипичное строение лица. У больных детей часто заметно снижен интеллект и нарушено психическое развитие. Они не успевают в школе, быстро утомляются, плохо воспринимают устную речь, редко заводят друзей, избегают общения с незнакомыми людьми. Психика пациентов лабильна: часто возникают периоды полного равнодушия к происходящим событиям, чередуются радость и печаль, настроение меняется по малейшему поводу. Мальчики плохо адаптируется в социуме. Одни становятся скромными, замкнутыми, тихими и апатичными, другие — импульсивными и резкими, у третьих появляются склонности к криминалу.
У подростков развивается андрогенный дефицит, который проявляется определенными симптомами:
- У больных увеличиваются грудные железы. Развивается двусторонняя, безболезненная и необратимая гинекомастия, сохраняющаяся пожизненно. Ранняя гормонотерапия снижает выраженность гинекомастии. Половые гормоны необходимо принимать сразу после постановки диагноза.
- У большинства детей обнаруживают патогномоничный признак заболевания – маленькие и плотные яички. При этом половой член имеет размеры, не соответствующие возрасту. Сформированная мошонка часто без складок и пигментации. Простата не обнаруживается при пальпации. Гипогонадизм и гипогенитализм развиваются у всех больных.
- Волосы отсутствуют на лице и груди, растут на лобке треугольником. Вторичные половые признаки появляются очень поздно. Ухудшаются показатели спермограммы. Недоразвитие гортани проявляется высоким голосом у больных мужчин.
- Крипторхизм или неопущение яичек – врожденная аномалия, которая часто имеется у детей с синдромом Клайнфельтера. Мошонка становится асимметричной, в ней отсутствуют яички, возникает ноющая боль в паху.
- Неспецифические симптомы патологии — одышка, бледность кожи, гипергидроз, «жар» в теле, «приливы», покраснение шеи, кардиалгия, аритмия, гипертензия, сменяющаяся гипотензией, нарушение сна и аппетита, депрессивные признаки, отсутствие интереса к жизни, гипотонус кожи.
- Возникают боли в костях в результате остеопороза.

Инволюция тестикул сопровождается потерей фертильности. У молодых мужчин 20-25 лет присутствуют редкие поллюции, эрекция, сохраняется половое влечение. Ближе к 30 годам андрогенный дефицит становится максимально выраженным, что проявляется снижением либидо, уменьшением яркости оргазма и развитием импотенции. Взрослые пациенты часто становятся алкоголиками, наркоманами, гомосексуалами, особенно в условиях стресса. Ослабление полового влечения, нарушение половой функции и бесплодие – наиболее частые признаки, с которыми больные люди приходят к врачу.
Лица с синдромом Клайнфельтера часто страдают коллагенозами, гипо- или гипертиреозом, заболеваниями глаз, имеют аномалии скелета и пороки сердца. Если лечение патологии не начать вовремя, могут развиться тяжелые осложнения и печальные последствия. У больных нарушается психо-эмоциональное состояние, формируется умственная отсталость, появляются суицидальные наклонности, злоупотребление крепкими спиртными напитками, нарушается толерантность к глюкозе, кости становятся хрупкими, усугубляются врожденные пороки сердца, появляются новообразования молочных желез.
Многие случаи синдрома Клайнфельтера. остаются недиагностированными. Это приводит к отсутствию лечения, снижению качества жизни, инвалидизации больных, развитию остеопороза и сердечно-сосудистых заболеваний.
Диагностические мероприятия

Пренатальная диагностика
Инвазивная пренатальная диагностика и последующее кариотипирование позволяют поставить правильный диагноз.
Существует две методики, с помощью которых можно получить материал для исследования:
- Биопсию хориона проводят с 9,5 по 12 неделю беременности.
- Амниоцентез – с 16 по 18 неделю.
Каждый из этих методов может выявить хромосомные аномалии с точностью до 99%.
Во время диагностического хирургического вмешательства получают ткани плода, из которых извлекают ДНК будущего ребенка. В лаборатории генетический материал исследуют на наличие хромосомных патологий.
- При биопсии специалисты прокалывают пункционной иглой переднюю брюшную стенку беременной женщины и извлекают ворсинки хориона с плаценты. Процедура контролируется аппаратом УЗИ.
- Амниоцентез — забор амниотической жидкости, содержащей генетическую информацию. Специальную иглу вводят в полость матки под контролем датчика аппарата УЗИ и собирают околоплодные воды.
Постнатальная диагностика
Диагностикой генетических заболеваний в постнатальном периоде занимаются эндокринологи, андрологи и генетики.
Специалисты начинают свою работу со сбора жалоб, анамнеза жизни и болезни. Они выясняют: время появления симптоматики, ее изменение, случаи генетических недугов в семье и в ближайшем поколении. Затем переходят к визуальному осмотру, проведению при необходимости пальпации, перкуссии и аускультации. Скудная клиническая картина синдрома Клайнфельтера не всегда позволяет вовремя диагностировать патологию и начать заместительную гормонотерапию. Диагностическим признаком заболевания являются тельца Барра, которые обнаруживают в клетках слизистой оболочки рта.
Исследование кариотипа позволяет поставить окончательный диагноз. Кариотипирование проводится всем бесплодным мужчинам с гинекомастией и мальчикам с умственной отсталостью.
Дополнительные диагностические методы:
- Ультразвуковое исследование мошонки позволяет определить размеры и структуру яичек.
- Ультразвуковое исследование сердца проводится с целью обнаружения врожденных пороков.
- Денситометрия — метод выявления остеопороза.
- Определение в крови половых гормонов — тестостерона, ФСГ и ЛГ.
- Спермограмма — анализ эякулята, проводимый с целью определения фертильности мужчины, наличия половых заболеваний, количества и активности сперматозоидов. У лиц с синдромом Клайнфельтера отмечается снижение количества или полное отсутствие сперматозоидов в эякуляте.
- Биопсия яичек выявляет состояние сперматогенеза и имеет большую диагностическую ценность.
Лечение
Синдром Клайнфельтера — неизлечимое патологическое состояние. Больным показана заместительное и симптоматическое лечение. Фармакотерапия направлена на полную нормализацию общего состояния больного, исчезновение основных проявлений, восстановление вторичных половых признаков.

Гормонотерапия
Заместительная гормонотерапия проводится пожизненно и заключается в назначении препаратов – синтетических аналогов тестостерона, которые вводятся внутримышечно, перорально или сублингвально. «Тестостерона пропионат», «Тестостерона энантат», «Сустанона-250», «Метилтестостерон» являются препаратами выбора. Наиболее популярны инъекционные формы тестостерона. Дозу лекарственного средства подбирает врач индивидуально каждому пациенту. При этом необходим постоянный контроль уровня тестостерона в крови. Своевременно начатое и адекватное лечение придает фигуре мужеподобность, улучшает внешний вид и общее самочувствие больного, возвращает его к полноценной жизни.
- «Тестостерона пропионат» необходимо вводить каждые 2-3 дня. Такая монотерапия практически не проводится.
- «Тестостерона ципионат» – эфир пролонгированного действия, который необходимо использовать каждые 7-14 дней.
- Комбинированные препараты содержат смесь нескольких эфиров тестостерона. Они являются очень популярными в нашей стране. Вводят их также внутримышечно. «Тестостерона капронат» имеет продолжительное действие — до месяца.
- В настоящее время фармакологическая промышленность разработала микрокапсулированные лекарственные формы гормона, которые действуют три месяца. Такие препараты имеют существенный недостаток: они вызывают значительные колебания тестостерона в крови, что негативно сказывается на самочувствии больных.
Симптоматическое лечение

- При необходимости больным назначают препараты, укрепляющие костную ткань, снижающие кровяное давление, нормализующие уровень глюкозы в крови.
- При умственной отсталости используют нейрометаболики и психостимуляторы.
- В подростковом возрасте назначаются пластыри на основе тестостерона.
- Увеличенные молочные железы удаляют операционным путем. Больным проводят мастэктомию. Криптохизм лечат также с помощью хирургического вмешательства. Операцию проводят детям в возрасте 1-1,5 лет. Неопущение яичка приводит к патологическим процессам в тканях семенных желез и развитию необратимого бесплодия.
- Психотерапия показана всем больным с целью повышения трудоспособности и социальной адаптации.
- Детям назначают занятия ЛФК, коррекционные занятия с логопедом, закаливание.
Современные методы экстракорпорального оплодотворения позволяют больным с синдромом Клайнфельтера иметь детей. При этом забор генетического материала осуществляют непосредственно из яичка путем его биопсии. Сперматозоидами оплодотворяют яйцеклетки и получают здоровое потомство. Донорская сперма — еще один шанс завести ребенка в семьях, где партнер болен.
Прогноз
Прогноз для жизни в целом благоприятный, а для восстановления способности к оплодотворению — крайне отрицательный. Больные способны вести нормальную жизнь, отдыхать и трудиться. Адаптироваться им в современном обществе поможет ранняя гормональная терапии и психотерапия.
Специфической профилактики заболевания не существует. Медико-генетическое консультирование проводится в обязательном порядке тем лицам, у которых в предыдущих поколениях регистрировались наследственные недуги. Правильное питание позволяет нормализовать массу тела и предупредить развитие ожирения и сахарного диабета.
Видео: о синдроме Клайнфельтера
Источник
Синдром Клайнфельтера: причины, клиника, диагностика и лечениеСиндром впервые был описан в 1942 г. Клайнфельтером с соавторами. В 1956 г. Планкетт и Барр обнаружили у мужчин с синдромом Клайнфельтера тельца полового хроматина в ядрах клеток слизистой оболочки полости рта, а Полани и Форд с сотрудниками показали, что у больных в хромосомном наборе имеется лишняя Х-хромосома. Этиология заболевания неизвестна. Возникновение патологического синдрома обусловлено неправильным расхождением половых хромосом в процессе овогенеза или сперматогенеза, а также на ранних стадиях эмбриогенеза, в результате чего нарушается нормальное развитие и функция половых органов. Синдром Клайнфельтера возникает у лиц с мужским фенотипом и характеризуется генетически полисомией по Х-хромосоме, Y-хромосоме или по X-и Y-хромосомам, а также мозаицизмом, при котором в одном из клеточных клонов выявляется полисомия по указанным выше хромосомам. Наиболее частым вариантом является 47XXY хромосомный комплекс. Описаны случаи заболевания, характеризующиеся 48XXXY, 49XXXXY, 48XXYY, 49XXXYY, 47XYY, 48XYYY, 46XX/47XXY, 46XY/47XXY, 45X/46XY/47XXY, 48XXXY/ 49XXXXY, 48XXXY/49XXXXY/50XXXXXY кариотипами. Несмотря на выраженные различия в хромосомном комплексе, клиническая картина синдрома Клайнфельтера в основном является общей для всех вариантов патологических изменений в половых хромосомах. Во всех случаях синдрома Крайнфельтера (за исключением полисомии по Y-хромосоме) у больных выявляется одно или более хроматиновых телец в ядрах клеток слизистой оболочки полости рта. Распространение заболевания среди населения, составляет 1:1100, среди олигофренов— 1:95, а среди мужчин, страдающих бесплодием, — 1:9. Клиническая картина синдрома характеризуется высоким ростом больных, евнухоидными пропорциями тела, наличием некоторых черт, присущих женскому типу строения туловища (широкий таз, узкие плечи, одно-или двусторонняя гинекомастия, отложение жира но женскому типу). Вторичные половые признаки — оволосение на лобке, под мышками, па лице — выражены слабо. Половой член, как правило, обычного размера, яички располагаются в мошонке, значительно уменьшены в размере, мягкие, иногда плотные. Половое чувство нередко у больных сохранено, но нормальный сперматогенез нарушен (азооспермия), что приводит к бесплодию. При гистологическом исследовании яичек выявляется склерозирующая гиалинизация семенных канальцев, уменьшение количества клеток Сертоли и гиперплазия клеток Лейдига. Выделение с мочой 17-кетостероидов, прегнандиола и прегнантриола находится на нижней границе нормы, выделение эстрогенов может быть несколько увеличенным. Обращает па себя внимание нормальное гистологическое строение яичек до пубертатного периода.
При исследовании функции других желез внутренней секреции отмечено увеличение продукции гормона роста и фолликулостимулирующего гормона передней доли гипофиза: функция щитовидной железы обычно не представляет отклонений от нормы. По данным различных авторов, у больных с синдромом Клайнфельтера нередко выявляется пониженная толерантность к глюкозе и клинический сахарный диабет. Обращает на себя внимание довольно большая частота нарушения интеллекта, вплоть до олигофрении. Указанные выше нарушения более выражены у больных с увеличением количества Х-хромосом более двух. При исследовании электроэнцефалограммы выявляется общемозговая патология. В отдельных случаях патология половых хромосом при синдроме Клайнфельтера сочетается с патологией соматических хромосом, что является результатом неправильного расхождения хромосом в процессе мейоза. Так, описаны сочетания синдрома Клейнфельтера и болезни Дауна, Диагноз и дифференциальный диагноз синдрома Клайнфельтера. Диагноз синдрома Клайнфельтера устанавливается на основании характерной клинической картины заболевания, включая обязательный признак его — гипогенитализм. Поскольку клиническая картина, свойственная синдрому Клайнфельтера, может наблюдаться при заболеваниях, не сопровождающихся патологией половых хромосом, диагноз требует подтверждения при помощи исследования полового хроматина, который всегда положителен при синдроме Клайнфельтера, или анализа хромосомного комплекса. В случаях подозрения на синдром Клайнфельтера, обусловленный полисомией по Y-хромосоме (гипогенитализм, высокий рост, увеличение нижней челюсти), вопрос о правильной диагностике может быть решен только после исследования кариотипа, так как половой хроматин при этом варианте синдрома будет отрицателен. Так как диагноз заболевания до полового созревания представляет большие трудности, то в основном обследованию подлежат все молодые люди, страдающие гипогенитализмом, при слабой выраженности вторичных половых признаков и гинекомастии. Обычно этот контингент больных направляют к эндокринологам для уточнения диагноза медицинские комиссии при райвоенкоматах. Это требует широкого внедрения метода исследования полового хроматина, так как другие методы диагностики дают мало информации для решения вопроса о правильном диагнозе. — Также рекомендуем «Синдром Шерешевского—Тернера у мужчин. Ложный мужской гермафродитизм» Оглавление темы «Аномалии половых хромосом и желез»:
|
Источник