Синдром кернса сейра тип наследования

Что такое синдром Кернса-Сейра?
Синдром Кернса-Сейра (англ. Kearns–Sayre syndrome, сокращённо KSS) — это редкое нервно-мышечное расстройство, которое обычно начинается в возрасте до 20 лет. Расстройство является результатом аномалий в ДНК митохондрий — маленьких стержнеобразных структур, обнаруживаемых в каждой клетке тела, производящие энергию, которая управляет клеточными функциями.
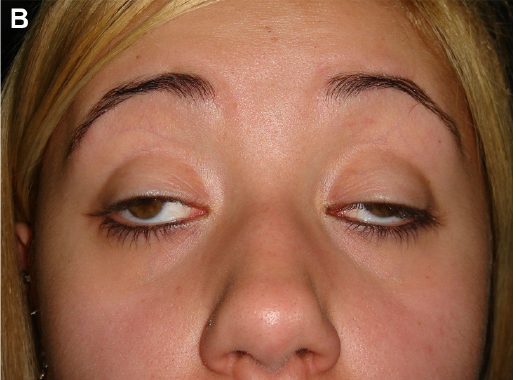
Митохондриальные заболевания коррелируют со специфическими мутациями ДНК, которые вызывают проблемы со многими органами и тканями организма. Синдром Кернса-Сейра характеризуется прогрессирующим ограничением движений глаз до полной неподвижности, сопровождающейся опущением века (см. фото). Патология также связана с ненормальным накоплением пигментированного материала на мембране, выстилающей глаза.
Дополнительные симптомы могут включать слабость скелетных мышц, блокаду сердца (дефект сердечной проводимости), низкий рост, потерю слуха, неспособность координировать произвольные движения (атаксия), нарушение когнитивной функции и диабет. Приступы нечасты. С KSS также могут быть связаны некоторые эндокринные расстройства.
Причины
Похоже, что большинство случаев синдрома Кернса-Сейра происходит в результате новой спонтанной делеции большого количества генетического материала, обнаруженного в ДНК митохондрий (мтДНК). Митохондрии, которые найдены сотнями в клетках тела, особенно в мышечной и нервной ткани, несут чертежи для регулирования производства энергии. В отличие от генетических инструкций клеточных хромосом (аутосомных ДНК), которые находятся в ядре каждой клетки, митохондриальные генетические инструкции находятся вне цитоплазмы в ядре клетки.
В крайне редких случаях эти делеции в митохондриальном генетическом материале могут наследоваться от матери. Генетические инструкции для митохондрий (мтДНК), обнаруженные в сперматозоидах, обычно разрушаются во время оплодотворения. В результате считается, что человеческая мтДНК происходит от матери. Пострадавшая мать может передать мутацию всем своим детям, но только дочери, а не сыновья передадут мутацию своим детям.
Как нормальная, так и мутированная мтДНК могут существовать в одной и той же клетке, и эта ситуация известна как гетероплазмия. Количество дефектных митохондрий может быть больше числа нормальных митохондрий. Симптомы синдрома Кернса-Сейра могут не появиться ни в одном конкретном поколении, пока делеция не затронет значительную часть митохондрий. Неравномерное распределение нормальной и мутантной мтДНК в разных тканях может влиять на разные органы у членов одного и того же семейства. Это может привести к различным симптомам у пострадавших членов семьи.
Симптомы
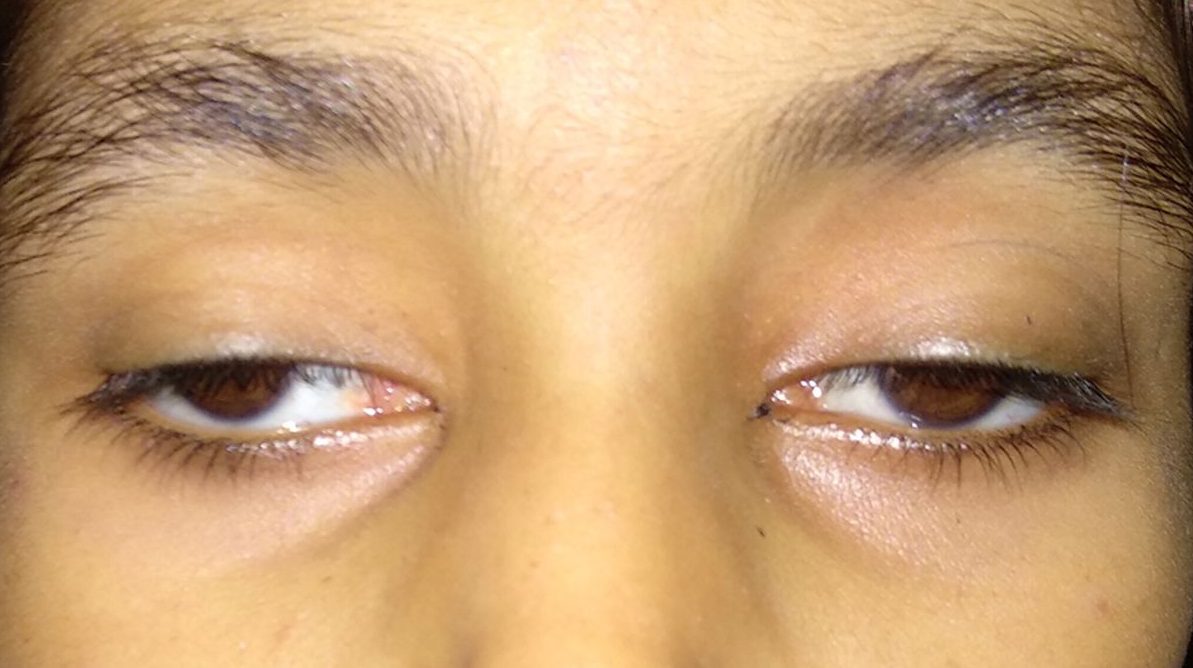
Тремя основными признаками KSS являются прогрессирующий паралич определенных глазных мышц (хроническая прогрессирующая внешняя офтальмоплегия); ненормальное накопление окрашенного (пигментированного) материала на богатой нервом мембране, выстилающей глаза (пигментный атипичный ретинит), приводящее к хроническому воспалению, прогрессирующей дегенерации и истощению определенных структур глаза (пигментная дегенерация сетчатки); и сердечные заболевания (кардиомиопатия), такие как блокада сердца. Симптомы этого расстройства обычно проявляются в возрасте до 20 лет.
В большинстве случаев первой физической характеристикой этого расстройства является замедление роста. Кроме того, понижение верхнего века (птоз) из-за слабости одной из мышц века также наблюдается в раннем детстве. Затем могут быть затронуты другие мышцы, участвующие в координации движений глаз, которые постепенно становятся слабее и в конечном итоге приводят к параличу определенных движений глаз.
В конце концов, мышечная слабость может распространиться на другие части лица, горла (глотки), шеи и/или плечи. Мышечная слабость в таких местах может мешать разговору и/или глотанию (дисфагия). По мере прогрессирования заболевания могут поражаться верхние части рук и ног, что приводит к прогрессирующему нарушению координированных движений (атаксии) и/или шаткой или прерывистой походке (титубации).
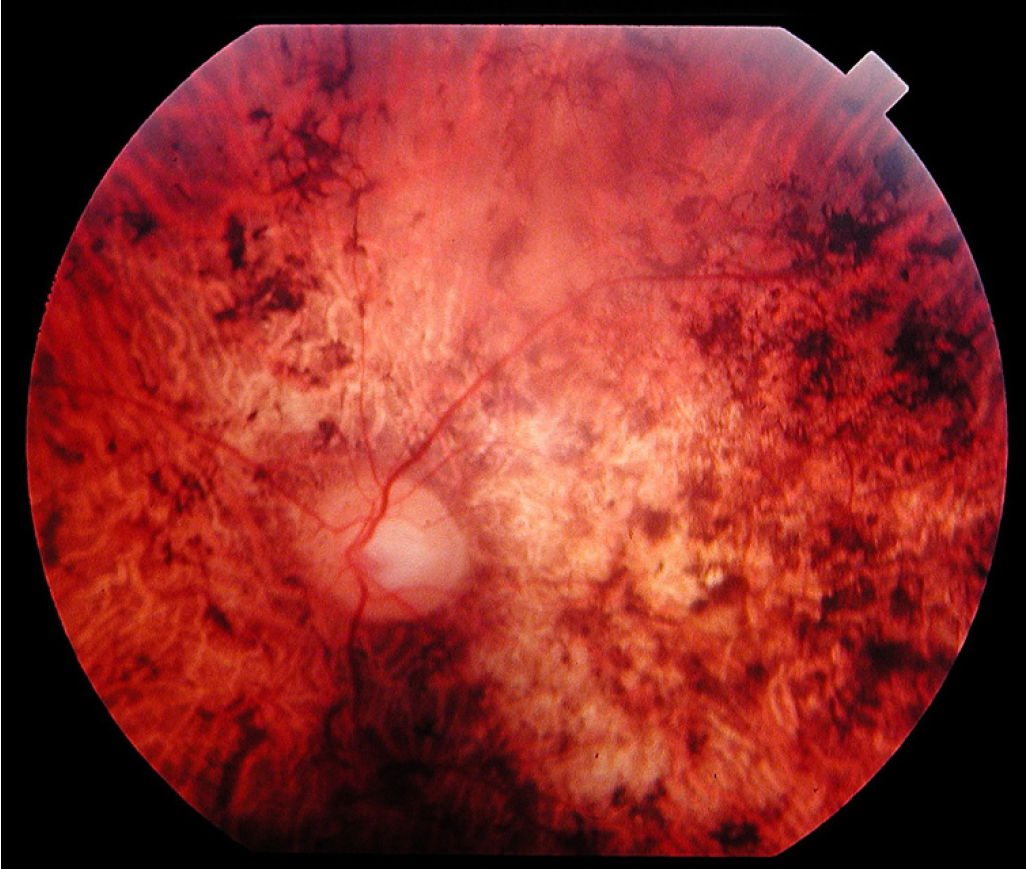
У большинства людей с синдромом Кернса-Сейра также будут проблемы со зрением из-за ненормального накопления окрашенного (пигментированного) материала на тонкой мембране, которая выравнивает глаза (пигментный атипичный ретинит) и прогрессирующей дегенерации определенных участков глаза (пигментная дегенерация сетчатки). Этот дегенеративный процесс может в конечном итоге повлиять на зрительный нерв (атрофия зрительного нерва), на слои мембран позади сетчатки (сосудистой оболочки) и/или на прочное белое наружное покрытие глазного яблока (склеру).
В некоторых случаях пострадавшие могут также испытывать ночную слепоту; быстрые непроизвольные движения глаз (нистагма); и снижение остроты зрения. В редких случаях аномальное помутнение передней части глазного яблока (роговицы) может также способствовать нистагму и снижению остроты зрения.
Третья первичная находка у людей с данным синдромом — это вмешательство в передачу нервных импульсов (проводимости), которые контролируют деятельность сердечной мышцы (блокада сердца). Серьезность таких нарушений проводимости может варьироваться по-разному среди пострадавших людей.
В редких случаях KSS также может быть связан с другими расстройствами или состояниями, включая отсутствие определенных рефлексов, почечные аномалии и/или периферическую нейропатию.
Периферическая нейропатия — это редкое заболевание, которое может поражать один или несколько нервов тела, вызывая боль и слабость. Периферическая нейропатия может влиять на сенсорную, моторную, рефлекторную или функцию кровеносных сосудов.
Диагностика
Диагноз KSS можно заподозрить, когда три основные характеристики, связанные с этим расстройством, возникают в связи друг с другом. К ним относятся паралич определенных глазных мышц (хроническая прогрессирующая внешняя офтальмоплегия), аномальная окраска тонкой мембраны, выстилающей глаза (пигментный атипичный ретинит) и другие изменения в структурах глаза (пигментная дегенерация сетчатки) и заболевания поражение сердца (кардиомиопатия), особенно нарушения проводимости (например, блокада сердца). Диагноз синдрома Кернса-Сейра может быть подтвержден тщательной клинической оценкой и различными специализированными тестами.
Такие специализированные тесты могут включать электрокардиограмму для выявления наличия и оценки тяжести блокады сердца, уровней молочной кислоты в крови и спинномозговой жидкости, биопсии мышц, чтобы продемонстрировать наличие характерных аномалий в мышечной ткани (рваные красные волокна), и/или позвоночника, чтобы определить, есть ли повышенные уровни определенных белков спинномозговой жидкости.
Биопсия мышц может определить наличие удаленной мтДНК, которая обычно не обнаруживается в образце крови. В некоторых случаях KSS уровни других веществ (например, сывороточная креатинкиназа, лактат крови, гамма-глобулин и/или пируват) могут быть повышены в крови.
Микроскопическое исследование образцов ткани биопсии под электронным микроскопом может выявить большое количество аномальных митохондрий в скелетной и глазной мышечной ткани. В некоторых случаях может использоваться компьютерная томография для выявления ненормального накопления кальция и/или поражений, поражающих определенные участки мозга.
Лечение
В настоящее время не существует эффективного способа лечения нарушений митохондрий при синдроме Кернса-Сейра. Лечение обычно симптоматическое и поддерживающее. Управление симптомами включает в себя несколько специальностей в зависимости от пораженных органов.
Наиболее важным является регулярное и долгосрочное наблюдение у кардиологов. Проблемы с сердечным импульсом, такие как сердечная блокада, можно лечить с помощью кардиостимулятора. Другие консультации могут включать аудиологию, офтальмологию, эндокринологию, неврологию и нейропсихиатрию.
Эндокринологические нарушения (например, сахарный диабет или гипопаратиреоз) можно лечить с помощью лекарств. В некоторых случаях лечение может включать заместительную гормональную терапию. Другие виды лечения будут зависеть от конкретных условий.
Хирургия может использоваться, чтобы исправить проблемы со зрением; в некоторых случаях, помогает ли операция улучшить зрение, часто зависит от того, насколько далеко продвинулись изменения сетчатки. Различные устройства могут помочь улучшить нарушения зрения у пострадавших людей. Конкретные используемые устройства и/или методы хирургического лечения будут зависеть от тяжести и конкретной комбинации имеющихся нарушений зрения.
Генетическое консультирование может быть полезным для пострадавших людей и их семей. Командный подход для детей с этим расстройством также может быть полезным и может включать специальную социальную поддержку и другие медицинские услуги, включая физическую и профессиональную терапию.
Прогноз
Синдром Кернса-Сейра — медленно прогрессирующее расстройство. Прогноз для людей с данным синдромом варьируется в зависимости от тяжести и количества вовлеченных органов. Ранняя диагностика и периодическая электрокардиограмма (ЭКГ) важны, поскольку блокада сердца может вызвать смерть у 20 процентов пациентов. Ранняя имплантация кардиостимулятора может принести большую пользу и увеличить продолжительность жизни у многих пациентов.
Источник
Болезни, обусловленные мутациями в митохондриальной ДНК, проявляются не при рождении, а во второй, реже – в первой декаде жизни.
В ряду генетических заболеваний числится Синдром Кернса–Сейра (Kearns–Sayre syndrome, KSS) — митохондриальная миопатия, поражающая глаза и мышцы пациентов. Неоднозначная динамика симптомов, их не одновременное проявление, а также отказ от биопсии и детальных генетических исследований осложняет точную постановку диагноза.
История появления диагноза
В 60-е годы доктора медицины Томас Кернс и Джозеф Сейр обратили внимание на три симптома, встречающиеся одновременно (нарушения зрения, пигментацию сетчатки глаза, нарушения проводимости нервных импульсов в сердце), описали несколько клинических случаев протекания болезни. Только в конце 80-х годов, после проведения исследований в области изменений в митохондриальных структурах стало понятно, как связанны между собой различные нарушения здоровья у одного пациента.
Синдром получил название по именам специалистов, впервые описавших характерную клиническую картину.
Дальнейшее изучение KSS показало, что симптоматика синдрома может быть более вариабельной (мышечная недостаточность в конечностях, слабоумие, нарушения в других сенсорных системах, общая задержка развития, низкий рост), но речь все равно идет о системном заболевании из-за нарушений в ДНК митохондрий.

Патогенез и общий характер симптоматики
Среди зафиксированных случаев (всего около 300 детально описанных), только в нескольких можно говорить о семейной наследственности (у родителей зафиксированы те же симптомы, что и у детей). Остальные — мутации на ранних сроках беременности, подлежащие диагностированию после 4 года жизни ребенка.
Симптомы при синдроме Кернса Сейра развиваются по нарастающей:
- уменьшение двигательной активности;

- значительное ухудшение четкости зрения;
- птоз одного или двух век;
- минимизация движений глазным яблоком до полного их отсутствия;
- брадикардия, аритмия;
- нарушения слуха, координации движений;
- проблемы с глотанием пищи и слюны (реже);
- атрофия мышц конечностей;
- прекращение работы одного из желудочков сердца и смерть.
Этиология болезни такова: «испорченный» геном митохондрий продолжает копироваться в искаженной форме при каждом делении клетки. Поскольку среди его главных функций – нормальный энергетический обмен, то начинают страдать те органы, где затраты энергии максимальны (нервные волокна, сенсорная система, мышцы). При отсутствии симптоматического лечения митохондриальной миопатии наступает смерть (средние показатели — до 24-36 лет).
Примечательно, что параллельно с «испорченными» клетками развиваются и работают те, у которых митохондрии в норме.
Генетический аспект
Митохондриальные проблемы передаются по материнской линии при значительном количестве измененных генов в органелле. На практике это означает, что у здоровой по виду женщины-носителя могут родиться и здоровые, и больные дети.
Наука пока не выявила точной закономерности развития синдрома. Понятно, что важным является первый триместр беременности, когда происходит активное деление клеток и закладка систем органов. Какой именно фактор влияет на более активное деление дефектных клеток, чем здоровых, пока не выявлено.
Синдром Кернса–Сейра встречается одинаково часто у мужчин и женщин разных рас.
Характерный клинический случай
Синдром Кернса–Сейра может иметь разные первоначальные проявления и дальнейшую клиническую картину. К примеру, у 25-летнего мужчины с выраженным птозом обеих век, атрофией мышц конечностей и спины, отсутствием движений глазными яблоками, пониженным зрением, болезнь проявилась в младшем школьном возрасте.
При физических нагрузках (бег) появлялись сильные головокружения, рвота, боль в животе. В 12 лет специалисты заметили птоз левого века, через 5 лет такая же участь постигла правое веко. При этом отмечалось двоение в глазах.
На фоне симптоматического лечения опущение уменьшилось, но препараты помогали все меньше, что сочли последствием привыкания. Для предотвращения полной слепоты рекомендовали укорочение век. С 23-летнего возраста начала развиваться тугоухость, прогрессирует общая мышечная слабость.
Нарушений в интеллектуальном развитии, работе сердца не обнаружено. Постоянное наблюдение у кардиолога и невропатолога дает возможность стабилизировать состояние, снизить интенсивность прогрессирования болезни.

Симптомы, характерные для синдрома Кернса–Сейра
Митохондриальная миопатия может развиваться и другим путем, когда при раннем выявлении болезни прослеживается быстрое ухудшение состояния без отзыва организма на лечение. Например, родители 5-летнего мальчика обратились к врачам с жалобами на одновременный птоз обеих век, нарушения движений глазными яблоками. При дальнейшем наблюдении выявили также нарушения сердечного ритма, психофизическое отставание в развитии, торможение полового созревания. За 6 лет наблюдений синдром прогрессировал до:
- координационных нарушений;
- затрудненного жевания и постоянного поперхивания;
- атрофии мимических мышц;
- атриовентрикулярной блокады;
- наджелудочковых экстрасистол.
Главным достижением лечения в данном случае стала временная нормализация сердечного ритма.
Динамика синдрома индивидуальна, зависит от количественных изменений в митохондриях, систематически принимаемых препаратов, а также от сопутствующих болезней.
Диагностический подход
Точное диагностирование заболевания возможно только при проведении биопсии мышц на предмет выявления патологии. Косвенными показателями болезни являются:
- совокупность симптомов и их динамика;
- завышенные уровни пировиноградной и молочной кислот в крови пациента;
- отсутствие отклика на прозерин, калимин;
- данные электрокардиографии;
- исследования на возможные нарушения в работе эндокринной системы (выявлены у 3% пациентов, существенно усугубляют протекание болезни).
Выявление болезни обычно занимает длительный промежуток времени ввиду медленного нарастания симптомов, неготовности родителей к проведению биопсии и детального генетического анализа.
Возможности современной медицины

Оперативное подрезание век
Синдром Кернса Сейра не поддается полному лечению на данном этапе развития медицины. Предполагается, что возможно купирование всех симптомов при стимуляции к делению здоровых клеток, постепенному замещению ими больных волокон.
Механизм подобной операции не разработан.
Экспериментальные операции показали, что спутниковые клетки могут способствовать регенерации мышечных волокон.
Пациент может рассчитывать на:
- вживление кардиостимулятора для предотвращения остановки сердца;
- введение коферментов для понижения уровня пировиноградной и молочной кислот (наблюдается улучшение подвижности глаза);
- укорочение век (временно улучшает зрение);
- использование слухового аппарата;
- нормализацию уровня гормонов.
Все названые меры снижают дискомфорт в быту, но не предотвращают полностью дальнейшее развитие мышечной слабости.
Последствия и прогноз
Митохондриальная миопатия имеет не оптимистический прогноз: прогресс мышечной слабости до остановки сердца или дыхания. При надлежащем лечении возможно существенное увеличение продолжительности жизни, но с признанной инвалидностью (2-3 группа).
В любом случае, речь идет о пожизненном наблюдении у кардиолога и невропатолога с постоянной корректировкой препаратов, их доз в зависимости от показателей анализов крови.
Профилактических методов нет, ввиду незначительной изученности пусковых механизмов.
Действенным методом уменьшения негативного влияния синдрома на качество жизни станет своевременное обращение к врачу, прохождение всего комплекса обследований и начало симптоматического лечения.
Перед запланированной беременностью родителям стоит пройти тест на определение возможных генетических отклонений у общего ребенка.
Источник
Пять лет назад на базе Морозовской детской больницы был создан единственный Городской референс-центр врожденных и наследственных заболеваний, генетических отклонений, орфанных и других редких заболеваний у детей и подростков. Сегодня их диагностику и лечение в столице уже можно сравнить с лучшими мировыми практиками. О том, как в Москве помогают маленьким пациентам с редкой патологией, газете «Московская медицина.
О том, как в Москве помогают маленьким пациентам с редкой патологией, газете «Московская медицина. Cito» рассказала руководитель центра, заведующая отделением наследственных нарушений обмена веществ Морозовской детской больницы, врач-генетик Наталья Печатникова.
— Наталья Леонидовна, какие болезни считаются редкими?
— К орфанным относятся те заболевания, которые имеют распространенность не более 10 случаев на 100 тысяч населения. Большинство редких заболеваний носят наследственный характер, в основе их развития — генные мутации. Раньше такие болезни практически не поддавались лечению и дети просто не доживали до взрослого возраста.
Ситуация в корне изменилась в начале XXI века, когда появились новые методы лабораторной диагностики, были разработаны лекарства, лечебное питание и методики лечения. Раньше наблюдение за пациентами с редкими заболеваниями велось в разных лечебных учреждениях. При развитии неотложных состояний они поступали в стационары, где врачи не знали их «специфики» и не могли в полной мере оказать им помощь. В июне 2015 года в Морозовской больницы впервые в Москве был организован региональный референс-центр, что позволило сосредоточить всю помощь детям с редкими заболеваниями на базе одного учреждения. Сегодня здесь наблюдаются более 1,5 тысяч детей.
— По какому принципу функционирует центр?
— Центр работает по принципу «одного окна». Дети и подростки с орфанными и другими редкими заболеваниями получают весь комплекс медицинских и диагностических мероприятий, проходят диспансерное наблюдение и обеспечиваются специализированными продуктами лечебного питания и лекарствами в одном месте. Кроме того, в экстренных ситуациях, при любых осложнениях таким детям в многопрофильном стационаре могут оказать всю необходимую помощь. На лечение в отделение наследственных нарушений обмена веществ, входящее в структуру центра, поступают дети с уже подтвержденным диагнозом по неонатальному или селективному скринингу. Также многих своих пациентов мы находим в стенах больницы. Ребенок может поступить в больницу по скорой, но, когда симптоматика не укладывается в привычную картину заболевания, проводится ряд дополнительных обследований и уточняется диагноз. Некоторые дети поступают к нам на обследование по направлению врача-генетика. В идеальном варианте мы должны выписать ребенка с установленным диагнозом и с расписанной терапией. Но так случается достаточно редко, генетическая диагностика длится порой месяцы, поэтому, когда клинически и лабораторно диагноз подтверждается, ребенку назначается терапия и он выписывается под амбулаторное наблюдение.
— Вы упомянули про скрининг. Что включают в себя эти методики? Как проводится диагностика детей с редкими заболеваниями?
— В России работает система неонатального скрининга на пять наследственных болезней. В Москве с 2018 года скрининг расширен, массовое обследование новорожденных проводится на 11 наследственных заболеваний. Кроме того, благодаря поддержке Правительства Москвы и ДЗМ, в 2016 году в столице появилась программа селективного скрининга на наследственные болезни обмена веществ. Исследования проводятся методом тандемной масс-спектрометрии. В роддоме у младенцев берут анализ — несколько капель крови из пятки. Капиллярная кровь наносится на специальный фильтр и помещается в аппарат. Для диагностики заболеваний из группы нарушения обмена веществ важно знать содержание в крови аминокислот и ацилкарнитина. Новые технические достижения позволяют из этого же фильтра выделить редукционные ферменты, что позволяет диагностировать другую большую группу заболеваний — лизосомные болезни накопления, такие как мукополисахаридоз. Для дальнейшей диагностики применяются стандартные методы: МРТ, УЗИ, ЭКГ, но есть определенная специфика изменений, которая может «подтолкнуть к мысли» о наличии определенных наследственных заболеваний. Эти догадки мы подтверждаем специфическими лабораторными тестами.
— Как вы можете оценить первые результаты введения расширенного неонатального скрининга?
— Самое главное, что теперь можно поймать болезнь еще в период новорожденности. Большинство таких заболеваний, к сожалению, неизлечимы, но своевременный старт терапии позволяет добиться компенсации нежелательных последствий. Мы продолжаем наблюдать за нашими пациентами. Многие из них уже не вызывают у нас опасений, растут и развиваются по возрасту. Хотя первые результаты не столь масштабны, но для нас каждый ребенок с редким заболеванием, у которого достигнута ремиссия болезни, — это наша большая победа.
— Какая группа заболеваний за прошедший период получила наибольший результат в плане лечения?
— Прежде всего, фенилкетонурия. Эта болезнь — пионер среди наследственных заболеваний обмена веществ, это самая многочисленная группа пациентов — в Москве зарегистрировано 360 детей с таким диагнозом. Сегодня они ничем не отличаются от здоровых сверстников и даже не считают себя больными. Конечно, им приходится соблюдать некоторые ограничения, связанные со специальной пожизненной диетой. А ведь фенилкетонурия — наследственное заболевание, и, когда его не умели лечить, большинство пациентов погибали в первые годы жизни.
Если говорить в целом о лечении детей с наследственными заболеваниями, то сегодня мы реже сталкиваемся с внезапными ухудшениями их самочувствия, и это заслуга не только врачей, но и родителей. Мы — одна команда. Семьи с больными детьми, которые прошли непростой путь проб и ошибок, оказывают огромную поддержку вновь выявленным пациентам и их родителям, что снимает с врачей огромную психологическую нагрузку. Пациентские сообщества доброжелательно принимают новичков, помогают советами, делятся информацией, и просто общаются. Это очень важно.
— Ваш прогноз на будущее. Появится ли в будущем возможность полностью излечивать такие заболевания?
— Когда мы работаем с детьми с наследственной патологией, то боремся с природой, пытаемся ее обыграть, нивелировать последствия генной ошибки. Будущее — за генной терапией, это одно из самых перспективных направлений в лечении наследственной патологии. Сегодня большинство исследований в этой сфере направлено на создание препаратов, которые могли бы устранить генную поломку, то есть воздействовать на первопричину заболевания. Однако надо понимать, что это достаточно серьезное вмешательство в организм человека, поэтому нужны длительные исследования. Пройдут годы, а возможно десятилетия, прежде чем этот метод начнет применяться повсеместно. Расширение диагностических и лабораторных возможностей позволит обнаруживать новые редкие болезни, которые сегодня неизвестны. В плане терапии мы ждем появления новейших таргетных препаратов, более точных и эффективных, воздействующих на причину заболевания. И, конечно, важнейший вопрос — доступность этих препаратов и возможность их применения в нашей практике.
Источник: https://мороздгкб.рф
Источник