Синдром элерса данлоса код мкб

Содержание
- Описание
- Дополнительные факты
- Причины
- Классификация
- Диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Синдром Элерса — Данлоса.
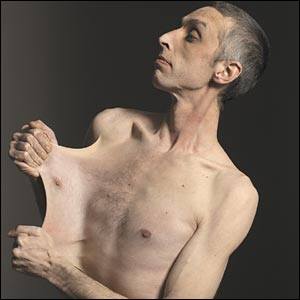
Гиперрастяжимость кожи при синдроме Элерса — Данлоса
Описание
Синдром Элерса. Данлоса — наследственная системная соединительнотканная дисплазия, обусловленная недостаточным развитием коллагеновых структур. В зависимости от клинического типа синдром Элерса-Данлоса может проявляться гипермобильностью суставов, необычайной ранимостью и растяжимостью кожи, склонностью к кровоизлияниям и кровотечениям, деформациями позвоночника и грудной клетки, миопией, косоглазием, птозом внутренних органов и пр. При диагностике синдрома Элерса-Данлоса учитываются клинические данные, результаты биопсии кожи и генотипирования; возможна пренатальная диагностика патологии. Лечение синдрома Элерса-Данлоса сводится к соблюдению щадящего режима, белковой диеты, симптоматической терапии.
Дополнительные факты
Синдром Элерса-Данлоса (несовершенный десмогенез, гиперэластическая кожа), наряду с несовершенным остеогенезом, синдромом Марфана и другими заболеваниями, относится к наследственным коллагенопатиям. Синдром Элерса-Данлоса неоднороден и включает в себя гетерогенную группу наследственных поражений соединительной ткани (соединительнотканных дисплазий), связанных с нарушением биосинтеза белка коллагена. Проявления синдрома Элерса-Данлоса носят системный характер и затрагивают опорно-двигательный аппарат, кожу, сердечно-сосудистую, зрительную, зубочелюстную и другие системы. Поэтому синдром Элерса-Данлоса представляет практический интерес не только для генетики, но и травматологии и ортопедии, дерматологии, кардиологии, офтальмологии, стоматологии.
Сложность верификации и наличие легких форм затрудняет получение точных сведений об истинной распространенности синдрома Элерса-Данлоса; частота диагностированных среднетяжелых случаев составляет 1:5 000 новорожденным, тяжелых форм — 1:100 000.
Гипермобильность суставов при синдроме Элерса — Данлоса
Причины
Различные варианты синдрома Элерса-Данлоса различаются по типу наследования, первичным молекулярным и биохимическим дефектам. Однако в основе всех клинических форм лежат мутации генов, обусловливающие количественную или структурную патологию коллагена. На сегодняшний день молекулярные механизмы синдрома Элерса-Данлоса установлены не для всех форм заболевания.
Так, известно, что I тип синдрома характеризуется снижением активности фибробластов, усилением синтеза протеогликанов, отсутствием ферментов, отвечающих за нормальный биосинтез коллагена. Синдром Элерса-Данлоса IV типа связан с недостаточностью продукции коллагена III типа; при VI типе заболевания имеет место недостаточность фермента лизилгидроксилазы, участвующего в гидроксилировании лизина в молекулах проколлагена. VII тип обусловлен нарушением превращения проколлагена I типа в коллаген; X тип — патологией плазменного фибронектина, участвующего в организации межклеточного матрикса.
Патоморфологическая картина при различных типах синдрома Элерса-Данлоса характеризуется истончением дермы, нарушением ориентации и потерей компактности коллагеновых волокон, разрастанием эластических волокон, увеличением числа сосудов и расширением их просвета.
Классификация
Всего выделяют 10 типов синдрома Элерса-Данлоса, различающихся по генетическому дефекту, характеру наследования и клиническим проявлениям. Рассмотрим основные из них:
I тип синдрома Элерса. Данлоса (классический тяжелого течения) — наиболее частый вариант заболевания (43% случаев) с аутосомно — доминантным типом наследования. Ведущим симптомом является гиперэластичность кожи, растяжимость которой по сравнению с нормой увеличена в 2-2,5 раза. Характерна гипермобильность суставов, носящая генерализованный характер, деформации скелета, повышенная ранимость кожи, склонность к наружным кровотечениям, образованию рубцов, плохому заживлению ран. У части больных выявляется наличие моллюскоподобных псевдоопухолей и варикозного расширения вен нижних конечностей. Беременность у женщин с I типом синдрома Элерса-Данлоса часто осложняется преждевременными родами.
II тип синдрома Элерса. Данлоса (классический мягкого течения) — характеризуется вышеописанными признаками, но выраженными в меньшей степени. Растяжимость кожи превосходит нормальную лишь на 30%; гипермобильность отмечается преимущественно в суставах стоп и кистей; кровоточивость и наклонность к рубцеванию незначительны.
III тип синдрома Элерса. Данлоса — имеет аутосомно — доминантное наследование, доброкачественное течение. Клинические проявления включают генерализованную повышенную подвижность суставов, скелетно-мышечные деформации. Остальные проявления (гиперэластичность и рубцевание кожи, геморрагии) минимальны.
IV тип синдрома Элерса. Данлоса — встречается редко, протекает тяжело; может наследоваться различными путями (доминантно или рецессивно). Гиперэластичность кожи незначительна, отмечается повышенная подвижность только суставов пальцев рук. Ведущим проявлением данного типа заболевания является геморрагический синдром: склонность к образованию экхимозов, спонтанных гематом (в т. Во внутренних органах), разрывам полых органов и сосудов (в т. Аорты). Сопровождается высокой летальностью.
V тип синдрома Элерса. Данлоса — имеет Х — сцепленное рецессивное наследование. Характеризуется повышенной растяжимостью кожи, умеренно выраженными гипермобильностью суставов, кровоточивостью и ранимостью кожи.
VI тип синдрома Элерса. Данлоса — наследуется по аутосомно — рецессивному типу. Кроме гиперэластичности кожи, наклонности к кровотечениям, повышенной подвижности суставов, имеются мышечная гипотония, тяжелый кифосколиоз, косолапость. Характерной чертой синдрома Элерса-Данлоса VI типа является глазной синдром, проявляющийся близорукостью, кератоконусом, косоглазием, глаукомой, отслойкой сетчатки.
VII тип синдрома Элерса. Данлоса (артроклазия) — наследуется как аутосомно — доминантно, так и аутосомно — рецессивно. Клиническую картину определяет низкий рост пациентов и гиперподвижность суставов, приводящая к частым привычным вывихам.
VIII тип синдрома Элерса. Данлоса — преимущественно наследуется аутосомно — доминантно. Ведущую роль в клинике играет хрупкость кожи, выраженный периодонтит, приводящий к ранней потере зубов.
X тип синдрома Элерса. Данлоса — характеризуется аутосомно — рецессивным наследованием; умеренной гиперэластичностью кожи и гипермобильностью суставов, стриями (полосовидной атрофией кожи), нарушением агрегации тромбоцитов.
XI тип синдрома Элерса. Данлоса — имеет аутосомно — доминантный тип наследования. У больных отмечаются рецидивирующие вывихи плечевых суставов, вывихи надколенника, встречается врожденный вывих бедра.
IX тип (Х-спепленный вариант вялой кожи) в настоящее время исключен из классификации синдрома Элерса-Данлоса. В современном варианте классификации синдрома Элерса-Данлоса рассматривается 7 основных типов заболевания:
• классический (типы I и II).
• гипермобильный (тип III).
• сосудистый (тип IV).
• кифосколиоз (тип VI).
• артроклазия (тип VIIB).
• дермоспараксис (тип VIIC).
• недостаток тенасцина-X.
Диагностика
Диагностика синдром Элерса-Данлоса проводится медицинским генетиком на основании генеалогических данных, анамнеза, клинического анализа, молекулярно-генетических исследований. Предварительно синдром Элерса-Данлоса может быть заподозрен при наличии больших диагностических критериев (гипермобильности суставов, гиперэластичности кожи, склонности к кровотечениям) и дополнительных малых (хрупкости кожи, патологии сердца, сосудов, глаз ).
Некоторые формы заболевания требуют проведения биопсии кожи для гистологического, гистохимического, электронно-микроскопического исследования.
Наличие в семье больных синдромом Элерса-Данлоса является показанием к медико-генетическому консультировании и проведению инвазивной пренатальной диагностики.
Больные с различными типами синдром Элерса-Данлоса могут нуждаться в наблюдении и обследовании детским травматологом-ортопедом, детским кардиологом, детским офтальмологом, детским стоматологом, сосудистым хирургом.
Лечение
Эффективная специфическая терапия синдрома Элерса-Данлоса не разработана. Детям требуется создание щадящего режима, исключающего излишнюю травматизацию суставов и кожи; ограничение физических нагрузок; соблюдение белковой диеты с включением в рацион костных бульонов, заливных блюд, студня. Обязательны регулярные курсы массажа, лечебной физкультуры, физиотерапии (магнитотерапии, электрофореза, лазеропунктуры).
Медикаментозная терапия синдрома Элерса-Данлоса включает применение аминокислот (карнитина), витаминов (С, Е, D, группы В), хондроитина сульфата, глюкозамина, минеральных комплексов (препаратов кальция и магния), метаболических препаратов (рибоксин, АТФ, коэнзим Q10) повторными курсами до1-1,5 мес. 2-3 раза в год.
При синдроме Элерса-Данлоса может быть показано хирургическое лечение: реконструкция грудной стенки, удаление псевдоопухолей, коррекция ВПС и пр.
Прогноз
На качество и продолжительность жизни больных синдромом Элерса-Данлоса влияет тип заболевания. Наиболее серьезный прогноз имеет IV тип синдрома Элерса-Данлоса – летальный исход может наступить вследствие разрывов сосудов, внутренних органов и кровотечений. Наличие синдрома I типа существенно ограничивает качество жизни. Относительно благоприятно протекание II—III типов болезни.
В целом, наличие синдрома Элерса-Данлоса сопряжено со множеством социальных трудностей, ограничивает полноценную физическую активность и выбор профессии.
Основные медуслуги по стандартам лечения | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Клиники для лечения с лучшими ценами
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Источник
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 26 июля 2015;
проверки требуют 20 правок.
Синдром Э́лерса — Данло́са (синдром Э́лерса — Данло́[2], син. Э-Д; англ. Ehlers-Danlos Syndrome, «гиперэластичность кожи» («Cutis hyperelastica»[3]), несовершенный десмогенез, несовершенный десмогенез Русакова, синдром Черногубова — Элерса — Данлоса) — это группа наследственных системных дисфункций соединительной ткани, вызванных дефектом в синтезе коллагена. В зависимости от отдельной мутации, серьёзность синдрома может измениться от умеренного до опасного для жизни. Лечения нет, но существует терапия (уход), смягчающая последствия.
Синдром назван в честь двух дерматологов, идентифицировавших его в начале XX века: Эдварда Элерса[en] (1863—1937) из Дании и Анри-Александра Данлоса[fr] (1844—1912) из Франции[4].
Симптоматика[править | править код]
Человек с син. Э-Д демонстрирует гиперподвижность суставов. |
Симптомы сильно варьируются в зависимости от типа болезни. Болезнь, как правило, поражает суставы, кожу и кровеносные сосуды, с симптомами, такими как свободные (плохо прикреплённые), сильно гнущиеся суставы; гладкая или эластичная, легко повреждающаяся кожа; неправильное заживление ран и формирование шрамов; маленькие и хрупкие кровеносные сосуды. Все формы затрагивают суставы, вызывая гиперподвижность, они выходят за пределы нормального диапазона движений.[5] В результате люди с «син. Э-Д» более подвержены различным травмам таким как: вывихи, подвывихи, растяжения связок, деформация и иногда разрыв мягких тканей. Так как синдром часто не диагностируется, некоторые случаи принимаются за жестокое обращение с детьми.[6]
Классификация[править | править код]
В прошлом было более десяти общепризнанных типов «син. Э-Д». В 1997 исследователи предложили более простую классификацию, которая сократила число главных типов до шести и дала им описательные имена.[7] Другие типы условно могут существовать, но о них сообщается только в отдельных семьях или они плохо характеризованы. За исключением типа гиперподвижности, были идентифицированы и отождествлены отдельные мутации — они могут быть точно определены генетическим анализом. Это особенно важно из-за значительной вариативности признаков в индивидуальном рассмотрении, из-за чего может быть перепутана классификация только на симптоматической основе.
По распространенности среди населения:
| Имя | Номер | Описание | OMIM | Ген(ы) |
| Гиперподвижность (Hypermobility) | тип 3 | Поражает 1 человека на 10 000 — 15 000, вызван аутосомным доминирующим механизмом. Возникает в результате мутации любого из двух генов, которые вызывают Сосудистый тип и син. Э-Д с дефицитом тенасцина-X, соответственно. Проявляется в гиперподвижности суставов; поражение кожи выражено меньше. Характерны хронические мышечно-скелетные боли. | 130020 | COL3A1, TNXB |
| Классический (Classical) | тип 1 и 2 | Поражает приблизительно от 2 до 5 человек на 100 000. Затрагивает коллаген типа V, также коллаген типа I. | 130000, 130010 | COL5A1, COL5A2, COL1A1 |
| Сосудистый (Vascular) | тип 4 | Поражает приблизительно 1 человека на 100 000. Вызван аутосомно-доминантным дефектом в синтезе коллагена типа III | 130050 | COL3A1 |
| Кифосколиоз (Kyphoscoliosis) | тип 6 | Аутосомно-доминантный дефект, вызывающий недостаток фермента, называемого Лизин Гидролаза[1]. Очень редок; описано немногим более 60 случаев. | 225400, 229200 | PLOD1 |
| Артрохалазия (Arthrochalasis) | типы 7A и B | Поражает коллаген типа I. Крайне редок, описано всего около 30 случаев. | 130060 | COL1A1, COL1A2 |
| Дерматоспараксис (Dermatosparaxis) | тип 7C | Также крайне редок, описано около 10 случаев. | 225410 | ADAMTS2 |
Тип гиперподвижность[править | править код]
Прежний тип 3, встречается у 1 человека на 10 000 — 15 000, что делает его самым распространённым вариантом болезни. Признаки и симптомы могут быть не диагностированы (не признаны) врачами или, как правило, ошибочно диагностированы как фибромиалгия и обычно больным не ставят диагноз, пока не проявятся серьёзные осложнения. Диагностика осуществляется, в основном, на клинических наблюдениях.
Основные признаки и симптомы включают в себя:
- Свободные, нестабильные суставы, подверженные: растяжениям, вывихам, подвывихам (частичный вывих), переразгибанию суставов
- Плоскостопие
- Высокое и узкое нёбо
- Лёгкие кровоподтёки
- Легко повреждающаяся бархатно-гладкая кожа
- Раннее начало остеопороза (обычно проявляется в середине 30 лет)
- Поражение сердца: в некоторой степени en:Dysautonomia или приобретённый порок сердца (такой как пролапс митрального клапана, что создаёт повышенный риск инфекции (эндокардит) во время операций (также возможно развитие до крайне опасной для жизни степени при пролапсе митрального клапана).[8]
Другие симптомы и осложнения могут включать в себя:
- Низкая плотность костей (остеопения) — предшественник остеопороза
- Мышечная слабость, часто усугубляется холодной погодой
- Деформации позвоночника, такие как: сколиоз (искривление позвоночника), кифоз (горб в грудном отделе), en:Tethered spinal cord syndrome, базилярная инвагинация (cranial settling), а также Мальформация Арнольда — Киари (поражение продолговатого мозга, мозжечка выраженные затылочными болями, нарушениями глотания, атаксией и другими симптомами)[9]
- Функциональные расстройства кишечника (функциональный гастрит, синдром раздражённого кишечника)
- Сдавление нервов (синдром запястного канала, парестезия, невралгия тройничного нерва)
- Болезнь Рейно
- Миалгия (боль в мышцах) и артралгия
- Чрезмерная усталость
- Преждевременный разрыв амниона (выкидыш) во время беременности en:Premature rupture of membranes
- Младенцы с гипервподвижностью суставов имеют слабый мышечный тонус (мышечная гипотония), который может задержать развитие таких моторных навыков как самостоятельное присаживание, вставание и хождение.
Боль, сопутствующая этому состоянию, является серьёзным осложнением.
Классический тип[править | править код]
При старой системе классификации он был разделён на два типа: тип I (тяжёлый) и тип II (умеренный). Поражает приблизительно от 2 до 5 человек на 100 000, и является вторым по распространённости. Поражает коллаген V и I типа. Важные симптомы затрагивают кожу и суставы. Больные как правило проявляют:
- гладкая, сильно эластичная, легко ранимая кожа
- уродливые или необычайно обширные шрамы, особенно на лбу, коленях, локтях и подбородке
- гиперподвижные суставы имеют тенденцию к вывихам, растяжению связок и подвывихам (обычно в коленной чашечке, в плече, в пястно-фаланговом суставе en:Metacarpophalangeal joint, и в височно-челюстном суставе
- Из-за сниженного мышечного тонуса, у младенцев может быть нарушено развитие моторных навыков
- Дети могут иметь склонность к развитию грыжи или смешению любого внутреннего органа.
В настоящее время не существует определённого теста для диагностики этого типа синдрома. И ДНК анализ и биохимические исследования используются для выявления пораженных болезнью. В некоторых случаях биопсия кожи была признана полезной при постановке диагноза. К сожалению эти тесты не достаточно надёжны, чтобы выявить всех больных. Если в семье есть несколько больных членов, то можно провести внутриутробную ДНК диагностику.
Сосудистый тип[править | править код]
Поражает приблизительно 1 человека на 100 000, вызван аутосомным доминантным дефектом в синтезе коллагена типа III. В старой системе классификации имел номер IV, этот тип является самой опасной разновидностью синдрома. Проведенные исследования определяют ожидаемую продолжительность жизни примерно в 48 лет. Тем не менее, эта цифра вероятно искажена и основана на факте, что этот тип (как все остальные типы синдрома) значительно не выявлен, и высокая пропорция смертельных случаев вызвана посмертным диагностированием. Повышение осведомлённости среди врачей и населения может помочь сделать эту цифру более точной, и сократить число преждевременных смертей.
Признаки и симптомы:
- Гиперподвижность, наиболее очевидна на пальцах рук и ног
- Хрупкие стенки сосудов оболочек органов и нежной кожи, имеют склонность к разрыву или образованию аневризмы
- Больные как правило имеют тонкую, бледную и прозрачную кожу (можно видеть вены на груди)
- Артериальная/кишечная/маточная хрупкость или трещины, разрывы
- обширные кровоподтёки
- Некоторые пациенты выражают характерные черты лица (большие глаза, маленький подбородок, тонкий нос и губы, мягкие уши) и имеют маленький рост
В результате возможности маточного разрыва, беременность может оказаться опасной для жизни. Доступно лабораторное тестирование. Кожная биопсия может служить доказательством аномальной структуры коллагена. Этот биомеханический анализ выявляет более 95 % случаев. Лабораторное тестирование рекомендуется лицам имеющим два или более значительных симптома. ДНК анализ может выявить изменения в пределах гена COL3A1. Эта информация может помочь при предродовой генетической консультации, когда один из родителей был диагностирован и известна его генетическая мутация или был продемонстрирован биомеханический дефект.
Тип кифосколиоз[править | править код]
В прежней классификации тип VI. Он очень редок, обнаружено немногим более 60 случаев, передаётся аутосомно-рецессивным механизмом. Основным симптом является общая нестабильность (непрочность) суставов. У младенцев наблюдается слабый мышечный тонус, задержка в развитии моторных навыков, прогрессирующее в течение жизни ненормальное искривление позвоночника сколиоз, при котором обычно больные не могут ходить к 20 годам. Легко ранимые глаза и кожа, также вероятна уязвимость кровеносных сосудов. Наблюдаются: спонтанная отслойка сетчатки, кровоизлияния в стекловидное тело, разрывы глазного яблока и роговицы, склер. Также у костей может быть снижена плотность.
Существует четыре основных медицинских критерия при диагностике этого типа. Это гиперподвижные суставы, слабый мышечный тонус у новорождённых, прогрессирующий с рождения сколиоз и хрупкие (уязвимые) глаза, что может придать голубоватый оттенок склерам или вызвать разрыв глаза.
Вызван изменениями в хромосоме 1 гена PLOD, который кодирует фермент лизил-гидролазу. Возможен лабораторный тест, в нём измеряется содержание в моче hydroxylysyl pryridinoline’а. Он крайне чувствителен и специфичен для данного типа синдрома, рекомендуется младенцам с тремя и более основными симптомами. Внутриутробный анализ применяется, если известно о существующем риске: диагностированы больные члены семьи, имеющие положительные результаты тестирования. Вышеупомянутый амниоцентез может быть выполнен, если зародышевые клетки берутся из амниотической жидкости и измерена активность фермента.
Артрохалазия[править | править код]
Характеризуется дефектом коллагена I типа за счет генов COL1.
При этом варианте ребенок может рождаться с врожденным вывихом бедра. Наследуется аутосомно – доминантно. Описано только около 30 случаев.[10]
Симптомы:
- Тяжелая генерализованная гипермобильность суставов с повторными вывихами (подвывихами)
- Гиперэластичная кожа;
- Атрофические шрамы;
- Мышечная гипотония;
- Кифосколиоз;
- Остеопения.[10]
Дерматоспараксис[править | править код]
Имеется дефектный ген ADAMTS2, о котором было сообщено в 10 случаях по всему миру.
Наследуется аутосомно – рецессивно.
Симптомы:
- Чрезвычайно хрупкая кожа, склонная к появлению синяков
- Раны и ссадины тяжело и долго заживают, образуя впоследствии атрофические шрамы
- Кожа на ощупь мягкая и податливая, ее слишком много и она образует складки
- Преждевременный разрыв плодных оболочек, грыжи (пупочные, паховые)[10]
Диагностика[править | править код]
Основана на данных:
- Анамнеза заболевания
- Обследования
- Биопсии кожи
- Молекулярно-генетических исследований
Дополнительно проводят:
- УЗИ внутренних органов
- Офтальмологические обследования
- Эхокардиограмму[10]
Лечение[править | править код]
Симптоматическое:
Для стабилизации деятельности сердечно-сосудистой и нервной системы, нормализации процессов в опорно-двигательном аппарате и коже используются:
- гемостатическая терапия — аскорбиновая кислота, этамзилат, антифибринолигики;
- витаминные препараты (витамины А, Е, В, С);
- препараты, стимулирующие обмен веществ и регенерацию;
- метаболические средства (карнитин-хлорид);
- минеральные комплексы;
- препараты, стимулирующие рост (инъекции соматотропного гормона);
- нейрометаболические стимуляторы, стимулирующие умственную деятельность.[10]
Профилактика[править | править код]
Основными направлениями профилактики неблагоприятных проявлений СЭД являются:
- правильно подобранная физическая нагрузка;
- предотвращение дислокаций;
- профилактические курсы лечения у офтальмолога, стоматолога;
- удаление псевдоопухолей;
- лечение патологии сердца, глаз.
См. также[править | править код]
Дисплазия соединительной ткани
Ссылки[править | править код]
- ↑ Disease Ontology release 2019-05-13 — 2019-05-13 — 2019.
- ↑ В русскоязычной литературе распространены оба названия, «Синдром Элерса — Данло́са» и «Синдром Элерса — Данло́». Большая медицинская энциклопедия (том 29, 1998), Геномная энциклопедия человека (1991), Малая медицинская энциклопедия (Т. 1, 1991) дают написание «Элерса-Данлоса».
- ↑ James, William; Berger, Timothy; Elston, Dirk (2005). Andrews’ Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. Page 512. ISBN 0-7216-2921-0.
- ↑ «Uncovered: How U.S. Health System Can Fail Even the Insured — A Woman Endures 16-Month Odyssey To Get a Diagnosis», John Carreyrou, Wall Street Journal, November 16, 2007
- ↑ Lawrence E.J. The clinical presentation of Ehlers-Danlos syndrome (англ.) // Advances in Neonatal Care (англ.)русск. : journal. — 2005. — Vol. 5, no. 6. — P. 301—314. — doi:10.1016/j.adnc.2005.09.006. — PMID 16338669.
- ↑ The Press Enterprise, Redlands mother stung by untrue suspicions presses for accountability in child abuse inquiries Архивировано 28 февраля 2009 года., 2008-04-03
- ↑ Beighton P., De Paepe A., Steinmann B., Tsipouras P., Wenstrup R.J. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK) (англ.) // American Journal of Medical Genetics (англ.)русск. : journal. — 1998. — Vol. 77, no. 1. — P. 31—7. — doi:10.1002/(SICI)1096-8628(19980428)77:1<31::AID-AJMG8>3.0.CO;2-O. — PMID 9557891.
- ↑ Ehlers-Danlos Syndrome, Hypermobility Type — GeneReviews — NCBI Bookshelf
- ↑ Sorry, but we cannot find this page. Дата обращения 10 апреля 2013.
- ↑ 1 2 3 4 5 Румянцев А.Г. Федеральные клинические рекомендации по диагностике и лечению синдрома Элерса-Данло у детей (2015).
Источник