Синдром эдварса код по мкб 10

Содержание
- Описание
- Симптомы
- Диагностика
- Лечение
Названия
Синдром Эдвардса.
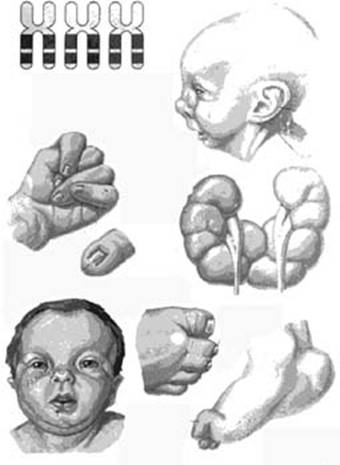
Симптомы при синдроме Эдвардса (трисомия 18)
Описание
Впервые трисомию по группе Е описали J. Edwads и соавт. (1960г. ). Частота синдрома Эдвардса среди новорожденных в среднем составляет 1:7000, замечено, что девочки поражаются в 3 раза чаще, чем мальчики. Ученым высказано предположение о стабилизирующем действии Х-хромосомы при абберациях пары 18, тогда как зиготы с трисомией 18, имеющие мужской генотип, элиминируются. Средний возраст матерей 32,5 года, отцов – 35 лет.
Симптомы
Длительность беременности превышает нормальную, и составляет в среднем 42 недели. Во время беременности диагностируют слабую активность плода и многоводие. Отмечается,что плацента имеет меньшие, чем обычно, размеры, часто обнаруживается только одна пупочная артерия; многие дети рождаются в асфиксии, с очень низкой массой тела и выраженной гипотрофией.
Фенотип больных с синдромом Эдвардса довольно характерен. Череп долихоцефалический, с низким лбом, затылок более широкий и выступающий. Часто встречается микроцефалия или гидроцефалия. Надглазничные валики сглажены, глазные щели узкие, наблюдается эпикант, птоз, часто встречается глазная патология, микрофтальмия, колобома, катаракта. Переносье вдавлено, но спинка носа тонкая, выступающая, ушные раковины расположены очень низко, диагностируется отсутствие мочки и козелка, недоразвиты завиток и противозавиток. Характерна микроретрогнатия. Рот мешьше обычных размеров, имеет треугольную форму, верхняя губа короче обычного. Небо высокое, иногда с расщелиной, шея короткая, нередко с крыловидной складкой.
Аномалии опорно-двигательного аппарата отличаются разнообразием: расширение шрудной клетки, укорочение грудины, таз узкий, деформации конечностей, ограничена подвижность в тазобедренных суставах, описаны вывихи бедра. Кисти и пальцы короткие, клинодактилия 5 пальцев кисти, дистально расположенный, гипоплазированный 1 палец кисти, сглажен тенар. Пальцы сжаты в кулак по типу «флексорной аномалии»: 2 и 5 пальцы расположены сверху и прикрывают прижатые к ладони 3 и 4 пальцы; 1 палец стопы широкий и короткий, синдактилия 2 и 3 пальцев. Типична для трисомии 18 форма стопы в виде своеобразной «качалки». Характерна общая мышечная гипотония. У мальчиков нередко встречается крипторхизм, гипоспадия; гипертрофия клитора у девочек.
Интеллектуальный дефект соответствует олигофрении в степени идиотии ил глубокой имбецильности и только в единичных описаниях мозаичного варианта трисомии 18 умственное недоразвитие менее грубое. Нередко у больного развиваются судороги.
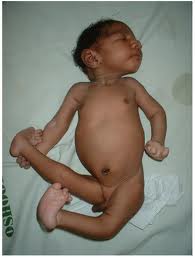
Внешний вид больного с синдромом Эдвардса (трисомия 18)
Диагностика
Дерматографическая картина при синдроме Эдвардса отличается своеобразными признаками: отмечается большая частота дуг на подушечках пальцев рук (примерно в 10 раз выше, чем в общей популяции), нередко дистальная сгибательная складка на пальцах не развита, у трети больных обнаруживается поперечная ладонная борозда, гребневый счет увеличен, осевой трирадиус обычно расположен дистально.
При аутопсии при синдроме Эдвардса находят разнообразные пороки развития практически всех органов и систем. С различной частотой встречаются аномалии ЦНС: аплазия или гипоплазия мозолистого тела, гипоплазия мозжечка, атрофия мозговых извилин. У 95% пациентов с трисомией 18 диагностируются пороки сердца и крупных сосудов, наиболее часто встречаются дефект межжелудочковой перегородки и незаращение артериального протока. Около половины всех случаев синдрома Эдвардса сопровождается аномалиями органов пищеварения: нарушение расположения кишечника, меккелев дивертикул, атрезия пищевода, атрезия заднего прохода. В 50% случаев отмечаются пороки развития мочеполовой системы – дольчатая или подковообразная почка, дупликация мочеточников, гипоплазия яичников.
При цитогенетическом обследовании в 80% случаев находят трисомию 18, у 10% больных – мозайцизм. Описаны случаи транслокационного варианта, двойной анеуплодии типа 48, ХХУ +18 с участием трисомного по хромосоме 18 клона.
Лечение
Нет патогенетического лечения синдрома Эдвардса на сегодняшний день, возможна лишь симптоматическая коррекция патологий. Прогноз для жизни неблагоприятный, средняя продолжительность жизни мальчиков 2-3 месяца, девочек – 10 месяцев. 30% больных умирают в течение 1-го года жизни, до года доживает лишь 10% больных. При мозаичных вариантах прогноз для жизни несколько лучше.
Источник
Рубрика МКБ-10: Q91.3
МКБ-10 / Q00-Q99 КЛАСС XVII Врожденные аномалии пороки развития, деформации и хромосомные нарушения / Q90-Q99 Хромосомные аномалии, не классифицированные в других рубриках / Q91 Синдром Эдвардса и синдром Патау
Определение и общие сведения[править]
Cиндром Эдвардса
Синонимы: трисомия 18
Трисомия по 18-й хромосоме встречается у новорожденных с частотой от 1:3300 до 1:10 000; у девочек бывает в 3 раза чаще, чем у мальчиков. Больные дети часто рождаются недоношенными или переношенными. Нарушения при трисомии по 18-й хромосоме гораздо тяжелее, чем при синдроме Дауна; лишь 50% пробандов доживают до 2-месячного возраста; 10% живут 1 год. Средняя продолжительность жизни мальчиков — 60, девочек — 280 дней.
Этиология и патогенез[править]
Большинство случаев связаны со свободной трисомией 18. Мозаичная трисомия 18 была обнаружена у нескольких пациентов с клинической картиной, которая варьируется от классической трисомии 18 до нормального фенотипа в зависимости от количества трисомных клеток, присутствующих в тканях. Фенотип трисомии 18, по-видимому, связан с наличием трех копий интервала 18q11-q12.
Клинические проявления[править]
Клиническая картина: череп необычной формы (узкий лоб и широкий выступающий затылок), низкое расположение ушей, микрогнатия, сгибательная контрактура кистей и стоп, дисплазия стоп, пороки сердца, сильная задержка психического развития. Главные нарушения обмена веществ и эндокринные расстройства: гипоплазия подкожной клетчатки, сильная задержка роста. Дисгенезия щитовидной железы или надпочечников встречается менее чем у 10% больных.
Синдром Эдвардса неуточненный: Диагностика[править]
Трисомию 18 можно заподозрить во время беременности по результатам ультразвукового исследования плода (задержка роста, пороки развития, множественные кисты сосудистого сплетения) и подтверждить кариотипическим анализом плода. Маркеры сыворотки (используемые для диагностики трисомии 21) также могут быть аномальными.
Дифференциальный диагноз[править]
Синдром Эдвардса неуточненный: Лечение[править]
Хирургическое лечение пороков развития мало способствует улучшению неблагоприятного прогноза, связанного с этим синдромом: 90% детей умирают в течение первого года жизни от сердечных, почечных или неврологических осложнений или от повторных инфекций.
Сообщалось о длительном выживании (в некоторых случаях до взрослого возраста), главным образом в случаях мозаичной или частичной трисомии (в результате транслокации). Большинство немозаичных пациентов имеют лишь ограниченную автономию (отсутствие речи и ходьбы).
Профилактика[править]
Прочее[править]
Источники (ссылки)[править]
Дополнительная литература (рекомендуемая)[править]
1. Buyse ML. Birth Defects Encyclopedia. Cambridge: Blackwell, 1990.
2. Emery A, Rimoin D. Principles and Practice of Medical Genetics (2nd ed). New York: Churchill Livingstone, 1990.
3. Gorlin RJ, et al. Syndromes of the Head and Neck (3rd ed). New York: Oxford University Press, 1990.
4. Hall JG, et al. Handbook of Normal Physical Measurements. New York: Oxford University Press, 1989.
5. Jones KL. Smith’s Recognizable Patterns of Human Malformation (4th ed). In M Markowitz (ed.), Major Problems in Clinical Pediatrics (vol VII). Philadelphia: Saunders, 1988.
6. McKusick V. Mendelian Inheritance in Man (10th ed.). Baltimore: Johns Hopkins University Press, 1992.
7. Rimoin DL. Disorders of the Endocrine Glands. In AA Dietz (ed.), Genetic Disease: Diagnosis and Treatment. Proceedings of the Fifth Arnold O. Beckman Conference in Clinical Chemistry. Monterey, CA: The Association for Clinical Chemistry, 1983.
8. Taybi H, Lachman RS. Radiology of Syndromes, Metabolic Disorders, and Skeletal Dysplasias (3rd ed). Chicago: Yearbook, 1990.
9. Vogel F, Motulsky AG. Human Genetics: Problems and Approaches (2nd ed). New York: Springer, 1986.
10. Wiedmann H-R, et al. Atlas of Clinical Syndromes—A Visual Aid to Diagnosis (2nd ed). St. Louis: Mosby, 1989.
11. Wynne-Davis R, Hall CM, Apley AG. Atlas of Skeletal Dysplasias. Edinburgh: Churchill Livingstone, 1985.
Действующие вещества[править]
Источник
Синдром эдвардса — это самое популярное генетическое заболевание после болезни Дауна. Исход недуга крайне неблагоприятен и выражен тяжелой олигофренией либо заканчивается ранней смертью. Потому так важно своевременно диагностировать имеющиеся нарушения и склонность к ним, чтобы предпринять правильные меры.
Причины и признаки
Синдром трисомия 18, как именуют еще патологию,представляет собой совокупность патологических состояний с неблагоприятным прогнозом для жизни. Всему виной появление 18 хромосомы. Кариотип больной девочки 47,XX, 18+ и тип мутации больного мальчика 47,XY, 18+.
Причины развития патологии:
- фактором возникновения служит образование лишней хромосомы в кариотипе при синдроме эдварса. Таким образом, вместо двух полноценных хромосом формируются три;
- 18 хромосома присутствует в организме родителя еще до зачатия;
- также наблюдается мозаицизм в некоторых случаях. Это означает, что не расхождение проявилось на раннем этапе развития зародыша.
Что конкретно стимулирует данное явление, ученым на данный момент неизвестно. Однако есть общие факторы риска, которые провоцируют возникновение генетических аномалий:
- возраст женщины старше 45;
- генетическая склонность;
- употребление наркотических веществ, злоупотребление спиртным и курение;
- продолжительный прием мощных медикаментозных препаратов;
- заболевания, передающиеся половым путем;
- радиация.
Перечисленные выше факторы являются только предполагаемыми, но не доказанными научно. Многие интересуются, какова частота встречаемости данной аномалии. На семь тысяч малышей приходится один ребенок с таким диагнозом. В связи с тем, что прогноз и качество жизни таких пациентов крайне негативные, целесообразно выявить патологию еще до оплодотворения.
Любопытно, что девочки с таким синдромом появляются на свет в три раза чаще, чем мальчики.
Симптомы
Семейной паре нужно понимать, что такое болезнь эдвардса, и какие страдания она способна принести родителям и ребенку. Симптоматика крайне не радостна. Первые признаки будут давать о себе знать уже после рождения:

- Асфиксия.
- Волчья пасть.
- Опущение век (птоз).
- Слабоумие.
- Нарушение глотания.
- Нарушение дыхания.
- Сердечные и сосудистые пороки.
- Заболевания системы пищеварения.
- Нарушения работы мочеполовой системы.
- Проблемы с психикой.
Учитывая все перечисленные симптомы, можно подвести итог, что больной ребенок никогда не станет полноценным членом общества, как бы этого не хотелось. Жизненный путь у него будет коротким, наполненным постоянными преградами. При этом больной не будет понимать и осознавать своего тяжелого состояния из-за тяжелой степени олигофрении.
Но это далеко не все проявления. Эдвардс описал свыше ста тридцати тяжелых симптомов данного недуга.
Диагностика
При выявлении аномалии в период вынашивания врачи рекомендуют делать аборт по медицинским показаниям. Потому диагностика имеет определяющее значение.
На УЗИ можно заметить такие проявления:
- многочисленные отклонения во внутриутробном развитии;
- многоводие;
- небольшой размер плаценты.
Это не явные признаки, а всего лишь предпосылки, которые служат серьезным поводом для проведения подтверждающих исследований.
Биохимический скрининг помогает выявить сывороточные маркеры.
Если риск появления на свет младенца с тяжелым заболеванием очень высок, тогда прибегают к таким диагностическим мерам:
- биопсия ворсин хориона;
- амниоцентез;
- кордоцентез;
- кариотипирование.
В случае если недуг не был диагностирован при беременности и больной ребенок родился, тогда он подлежит всестороннему обследованию.
У больного ребенка, появившегося на свет, обнаруживают пороки в развитии. Он проходит разных специалистов: кардиологов, хирургов, ортопедов и т.д.
После рождения малышу назначаются УЗИ почек и брюшной полости.
Беременность при данной патологии ничем не отличается от здоровой. Дополнительных сигналов о том, что это больной ребенок, нет. Потому определить наличие патологии возможно только с помощью применения современной диагностической техники.
Лечение
Такие пороки и аномалии, как синдром Эдвардса, к огромному сожалению, не сопоставимы с жизнью. Терапия в этом случае носит только симптоматический характер:

- поддержание жизненно важных функций;
- улучшение качества жизни;
- зондовое питание в первые дни после рождения;
- ИВЛ из-за трудностей дыхания;
- профессиональный пожизненный уход;
- систематическое наблюдение медиков.
Стоит сказать, что это такой недуг, который требует недюжинного терпения и сил.
При легком течении и благоприятном прогнозе возможно проведение хирургических коррекций. Прописываются медикаментозные средства для опорожнения кишечника.
Специфических методик профилактики этого заболевания нет. При наследственной предрасположенности необходимо обязательно проконсультироваться у генетика и пройти обследование.
Прогноз
Как уже было сказано, исход заболевания крайне неутешителен. Летальность составляет около 90% в течение первого года. Выжившие дети мучаются от многочисленных пороков и умственной и физической отсталости.
При легкой мозаичной форме прогноз более приятен, тем не менее,патологий очень много и они заметны окружающим.
Пациенты с таким диагнозом не способны к самообслуживанию и требуют систематического наблюдения. Если больной способен распознавать близких людей, то с ними он может по минимуму общаться. Иногда дети могут сами держать голову и кушать.
Кроме всего прочего больные входят в повышенную группу риска по заболеваемости раком почки. Велика вероятность возникновения отитов, синуситов и прочих ЛОР заболеваний.
Источник
Синдром Эдвардса – генетическая патология, которая характеризуется трисомией (дублированием) 18 хромосомы и сопровождается рядом пороков развития.
Общая информация
Трисомия по 18 хромосоме (синдром Эдвардса) – ненаследственное заболевание, мутация генов происходит случайно. Патология встречается в 1 случае из 3000 зачатий и 6000-7000 рождений. У девочек она диагностируется чаще, чем у мальчиков в 3 раза.
Риск синдрома Эдвардса повышается с возрастом матери – после 45 лет он составляет 0,7%. Болезнь негативно отражается на течении беременности: отмечается многоводие, недостаточная активность плода и роды позже срока (на 42-43 неделе).
60% детей с синдромом Эдвардса умирают в период внутриутробного развития. Продолжительность жизни 2-3 месяца – для мальчиков, 10 месяцев – для девочек. Смерть наступает из-за серьезных нарушений в работе внутренних органов.
Причины
Причина синдрома Эдвардса – присутствие в наборе хромосом (кариотипе) «лишней» 18 хромосомы. В норме зигота (яйцеклетка после оплодотворения) должна иметь 23 пары хромосом, полученных в результате слияния женской и мужской гамет (половых клеток). Под влиянием не установленных наукой факторов происходит дублирование генетического материала. У зиготы вместо двух 18 хромосом появляется три: материнская, отцовская и дополнительная.
Мутация может осуществиться как до оплодотворения, так и после него – на этапе деления яйцеклетки. Кариотип при синдроме Эдвардса имеет три варианта:
- полная трисомия (95% случаев) – три 18 хромосомы в каждой клетке плода;
- транслокационная (2%) – две 18 хромосомы и часть третьей, добавившаяся к другой хромосоме;
- мозаичная (3%) – три 18 хромосомы в некоторых клетках будущего ребенка.
Первая форма трисомии синдрома Эдвардса характеризуется более сложным течением, чем две другие.
Симптомы
Симптомы синдрома Эдвардса варьируются в зависимости от условий развития эмбриона и формы трисомии. В большинстве случаев имеют место следующие внешние признаки:
- Масса тела при рождении – 2,1-2,2 кг.
- Искажение черепа – маленький размер, долихоцефалическая (удлиненная к затылку) форма, гидроцефалия.
- Деформация лицевых костей – узкий лоб, широкий затылок, недоразвитие нижней челюсти, маленький рот, высокое небо, нарушенный прикус, узкие глазные щели, углубленная переносица, суженный нос.
- «Заячья губа», «волчья пасть».
- Укороченная шея с кожной складкой.
- Низко посаженные уши неправильной формы – без козелка, вытянутые горизонтально, с узким слуховым ходом.
Синдром Эдвардса характеризуется значительным изменением опорно-двигательного аппарата. Обычно наблюдаются такие нарушения, как:
- Дисфункция суставов – они не могут нормально сгибаться и разгибаться.
- Дисплазия стоп – провисание свода с одновременным выступанием пятки («стопа-качалка»).
- Подвижность тазобедренных суставов.
- Расширение и укорочение грудной клетки
- Увеличение количеств дуг палацев рук, отсутствие сгибательной складки.
- Сращивание пальцев стопы, появление перепонок между ними.
При синдроме Эдвардса отмечаются множественные аномалии в работе внутренних органов, в частности:
- Пороки сердца – незакрытый артериальный проток, дефект перегородки между желудочками и так далее.
- Эндокринные проблемы, под влияние которых замедляется рост и недостаточно формируется подкожная клетчатка.
- Гипотония мышц.
- Аномалии пищеварительной системы – гастроэзофагеальныйрефлюкс, слабость сосательного рефлекса, атрезия некоторых участков ЖКТ, дивертикулы.
- Мочеполовые патологии – крипторхизм, недоразвитость яичников, деформация почек, удвоение мочеточников.
- Нарушение зрения.
 Ребенок с синдромом Эдвардса
Ребенок с синдромом Эдвардса
Каждый из признаков синдрома Эдвардса встречаются в 70-95% случаев. Абсолютно у всех детей есть проблемы с развитием головного мозга – умственная отсталость (вплоть до имбецильности).
Диагностика
Диагностику синдрома Эдвардса можно осуществить во время беременности Наиболее доступной методикой является ультразвуковое исследование.
Косвенные признаки синдрома Эдвардса на УЗИ на раннем этапе (12 недель):
- большой объем околоплодной жидкости;
- снижение частоты сердцебиения;
- брюшная грыжа;
- отсутствие носовых костей;
- 2 артерии в пуповине;
- кисты сосудистых сплетений.
На поздних сроках (третий триместр беременности) синдром Эдвардса на УЗИ может проявляться такими симптомами, как:
- аномалии костей головы и мягких тканей;
- пороки опорно-двигательного аппарата, сердца, сосудов, мочеполовой системы и так далее.
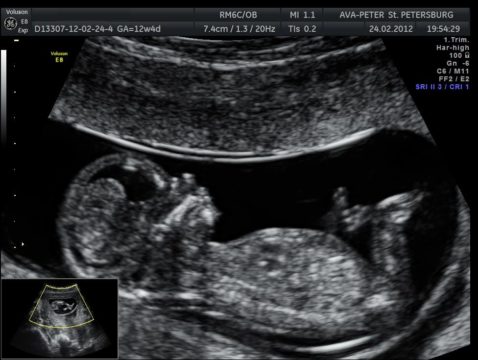 УЗИ – один из методов, позволяющих предположить синдром Эдвардса
УЗИ – один из методов, позволяющих предположить синдром Эдвардса
Более точным способом диагностики является биохимический анализ крови на 11-13 неделе беременности. Исследование предполагает определение уровня плазменного протеина А (РАРР-А) и хорионического гормона (β-ХГЧ). Затем полученные результаты соотносят с возрастом будущей мамы и сроком вынашивания ребенка. При отклонении показателей от нормы ставят высокий риск синдрома Эдвардса. Диагноз на этом этапе не определяется.
Затем осуществляется определение статуса плода с помощью различных методик:
Инвазивные способы выявления синдрома Эдвардса:
- Биопсия ворсин хориона (БВХ) – исследование небольшого образца плаценты. Доступна на сроке 8-12 недель.
- Амниоцентез – проникновение через амниотическую оболочку и взятие пробы околоплодных вод. Делается на сроке 14-18 недель.
- Кордоцентез – забор пуповинной крови. Проводится в 20 недель.
Все три метода дают возможность определить кариотип ребенка и подтвердить либо опровергнуть диагноз. Их недостаток – риск инфицирования плода и прерывания беременности.
К неинвазивным методикам относится выделение ДНК ребенка из крови матери.
Точность генетического тестирования очень высока: синдром Эдвардса подтверждается часто – в 90% случаев. Сложнее всего установить мозаичную форму заболевания: в проверяемый образец могут не попасть клетки с измененным кариотипом.
После рождения ребенка патология диагностируется на основании внешнего осмотра и кариотипирования – исследования крови с целью определения хромосомного набора.
Лечение
Причины возникновения синдрома Эдвардса заключаются в генетической мутации, потому избавиться от этого заболевания невозможно ни до, ни после рождения.
Лечение синдрома Эдвардса сводится к максимальному устранению аномалий строения. Но из-за высокой смертности в первые месяцы жизни и наличия множественных нарушений в работе всех систем хирургическое вмешательство в большинстве случаев оказывается нецелесообразным. Как правило, помощь ребенку с синдромом Эдвардса заключается в:
- восстановлении проходимости пищевода и кишечника;
- кормлении через зонд либо применении специальных смесей;
- использовании слабительных и пеногасителей;
- создании стерильных условий, поскольку дети с синдромом Эдвардса крайне подвержены различным инфекционным заболеваниям.
При относительно легкой форме патологии после достижения ребенком 2-3 летнего возраста становится возможным проведение операций по устранению косметических дефектов («заячьей губы», сросшихся пальцев), а также различных аномалий внутренних органов (порока сердца, грыж и так далее). Кроме того, требуется постоянная коррекция задержки психического развития с помощью обучающих методик.
Синдром Эдвардса увеличивает риск возникновения ряда заболеваний, среди которых:
- рак почек (опухоль Вильмса);
- пневмония;
- конъюнктивит;
- мочеполовые инфекции;
- фронтит, отит;
- апноэ по ночам;
- сердечно-сосудистые патологии так далее.
Пациенты с трисомией 18 хромосомы нуждаются в регулярных медицинских обследованиях.
Прогноз и профилактика
Синдром Эдвардса характеризуется неблагоприятным прогнозом: 60% больных умирают до трехмесячного возраста, еще 30-35% – до года. Выжившие 5-10% детей страдают от умственной отсталости и различных заболеваний, связанных с аномалиями строения.
При легкой форме мозаичного синдрома Эдвардса психическое и физическое развитие детей можно корректировать: научить их общаться с ограниченным кругом людей, привить некоторые навыки самообслуживания.
Поскольку патология относится к случайным хромосомным мутациям, предупредить его возникновение невозможно.
Источник