Синдром холт орама что это

Синдром Холта-Орама, также называемый синдромом «сердце-рука», является наследственным заболеванием, которое характеризуется нарушениями верхних конечностей и сердца. Холт и Орам впервые описали это состояние в 1960 году при обследовании пациентов с дефектами межпредсердной перегородки и аномалиями большого пальца.
Синдром Холта-Орама. Причины
Синдром Холта-Орама является генетическим расстройством, которое имеет аутосомно-доминантный признак.
- Дефектный ген находится на длинном плече хромосомы 12.
- Молекулярно-генетические исследования показывают, что болезнь вызывается мутациями, которые инактивируют фактор транскрипции TBX5.
- Спорадическая болезнь может развиваться De Novo при мутации в гене TBX5.
Синдром Холта-Орама. Патофизиология
Синдром наследуется по аутосомно-доминантному типу. Заболевание связано с мутациями в гене транскрипционного фактораTBX5, что в конечном итоге выливается в развитие аномалий сердца и верхних конечностей. Патофизиологические последствия являются прямым результатом развития пороков развития сердца и верхних конечностей. Информация по экологическим факторам, которые могут нести ответственность за развитие синдрома, не поступала.
Синдром Холта-Орама. Симптомы и проявления
Верхние конечности.
Хотя клинические проявления показывают переменный характер, аномалии верхних конечностей у детей присутствуют всегда. Аномалии могут быть односторонними или двусторонними, асимметричными и симметричными. Аплазия, гипоплазия, или другие аномальные проявления в этих костях проявляют целый спектр фенотипов (от простых аномалий и до полного отсутствия пальцев). Иногда порок развития конечностей может быть настолько серьезным, что у ребенка может развиться фокомелия (порок развития, при котором будут отсутствовать проксимальные участки рук). Наиболее распространенным проявлением синдрома являются пороки развития или слитые кости запястья. Кистевые аномалии являются единственными типами проявлений этого синдрома, которые присутствуют у каждого пациента с синдромом. Однако эти аномалии, у некоторых людей, могут быть обнаружены только при прохождении рентгенологических процедур.
Сердце.
Примерно 75% больных имеют некоторую сердечную аномалию. У большинства пациентов присутствует либо дефект межпредсердной перегородки либо дефект межжелудочковой перегородки, эти дефекты могут варьироваться в размере и расположении. Сердечные аномалии также могут включать в себя сердечные дефекты проводимости и фибрилляции предсердий. Эти аномалии часто присутствуют даже в отсутствие дефектов перегородки.
Синдром Холта-Орама. Фото

Полидактилия всех четырех конечностей.

Обратите внимание на мизинцы.

Синдром Холта-Орама у матери.

И у её сына.

Гипоплазия конечностей.

Слева рентген грудной клетки до операции. Правый желудочек, правое предсердие и центральная часть легочной артерии расширенна. Справа, рентген грудной клетки после оперативного закрытия дефекта межпредсердной перегородки, у пациента наблюдается значительное сокращение правого предсердия, желудочка и сокращение растяжения легочной артерии.

Проявления синдрома Холта-Орама.

Пупочная грыжа с полидактилией левой верхней конечности.

Рентген грудной клетки показывает кардиомегалию.

На снимке виден дополнительный палец.
Физическое обследование
- Деформации верхних конечностей
- Всегда присутствуют, но могут быть односторонними или двусторонними
- Левосторонние аномалии часто более тяжелые, чем аномалии правой руки
- Неравные длины рук происходят в связи с аплазией, гипоплазией, слиянием или аномальным развитием радиальной кости, запястья
- Аномалии предплечья
- Могут отсутствовать пальцы
- Аномалии большого пальца
- Ограничения в движениях плечевым суставом
- Фокомелия
- Поражения сердца
- Брадикардия
- Нерегулярный пульс (эктопия)
- Мерцательная аритмия
- Легочный систолический шум
- Голосистолический шум
Синдром Холта-Орама. Диагностика
- Рентгенография костей рук
- Рентгенография грудной клетки. Результаты могут продемонстрировать увеличенные легочные артерии в связи с развитой легочной гипертензией или кардиомегалией, также по результатам можно найти свидетельства застойной сердечной недостаточности.
- Эхокардиография. На этой процедуре можно определить присутствие дефектов перегородки или другие сердечные аномалии. Наиболее распространенной сердечной аномалией является дефект межжелудочковой перегородки. Некоторые пациенты также могут иметь ВСД. Необъяснимое, значительное расширение правого предсердия у плода может означать наличие синдрома Холта-Орама.
- ЭКГ для определения участия проводящей системы.
- Генетическая оценка также имеет важное значение — мутации в гене TBX5 обнаруживаются у около 75% лиц, отвечающим строгим клиническим критериям синдрома Холта-Орама.
Синдром Холта-Орама. Лечение
Медикаментозное лечение пациента может выполняться, как правило, как в амбулаторных, так и в стационарных условиях, хирургическое вмешательство также может быть необходимым. Пациентам с блокадой сердца может потребовать установка постоянного кардиостимулятора. Хирургическое вмешательство может быть выполнено для коррекции пороков сердца или, возможно, для улучшения функций конечностей.
Хирургическое вмешательство.
Большинство пороков сердца, такие как дефект межпредсердной перегородки либо дефект межжелудочковой перегородки поддаются хирургической коррекции, если у пациента не наблюдается легочная гипертензия или коллапс желудочка. Сегодня доступно несколько хирургических методов, которыми можно исправлять сердечные аномалии, к ним также можно отнести имплантацию чрескожных транскатетерных устройств, которыми можно закрыть отверстие в перегородке.
- Септальные дефекты без гемодинамически значимых аномалий не требуют коррекции.
- Дети с тяжелыми аномалиями конечностей могут быть направлены к ортопедами для рассмотрения и проведения специальных коррекционных процедур.
- Дети с укороченными конечностями могут извлечь выгоду из протезов.
Синдром Холта-Орама. Осложнения
- Сердечная недостаточность
- Аритмия
- Блокада сердца
- Мерцательная аритмия
- Инфекционный эндокардит
- Внезапная смерть
Синдром Холта-Орама. Прогноз
Прогноз, как правило, хороший, но он сильно зависит от тяжести сердечных пороков.
Источник
Синдром Холта Орама влияет на кости рук и сердце. Люди с расстройством имеют по крайней мере одну кость в запястье, которая не образуется (развивается) нормально. Другие кости в руках, плече могут также развиться ненормально.
Многие из этих изменений развития возможно увидеть только на Рентгеновском снимке. Также возникают проблемы с сердцем врожденные или приобретенные.
Синдром Холта-Орама вызван генетическими изменениями (патогенными вариантами, мутациями) гена TBX5. Наследуется аутосомно доминантным способом.
Диагноз может быть заподозрен, если у человека присутствуют характерные признаки. Рентген рук, запястий, эхокардиограмма сердца, генетическое тестирование используются для подтверждения. Варианты лечения включают операции для устранения проблем с костью или сердцем, а также физиотерапию.

Другие имена:
- Синдром сердца;
- HOS;
- Atriodigital дисплазия.
Симптомы
Признаки и симптомы Холт-Орама включают врожденные дефекты руки, запястья, сердца. Люди имеют по крайней мере одну из запястных костей, которая аномально сформирована. Другие кости верхних конечностей также могут образоваться аномально.
Они могут включать в себя отсутствующий, длинный большой палец, отсутствующие кости предплечья, плеча, проблемы с формой воротничной кости или лопаток. Иногда аномалии видны только на рентгеновском снимке.
Проблемы с сердцем
Около 75% имеют проблемы с сердцем. Наиболее распространенными проблемами являются отверстия в стенах камер сердца. Эти проблемы известны как дефекты предсердной перегородки (ASD), дефекты межжелудочковой перегородки (VSD) в зависимости от точного местоположения отверстия.
Сообщалось о других сердечных патологиях, включая патентный артериальный проток (PDA). Бывает, что сердечные дефекты, не вызывают никаких проблем или дают такие симптомы, как затрудненное дыхание, легкое утомление (усталость), высокое кровяное давление в артериях легких (легочная гипертензия). Часто, они опасны для жизни.
Некоторые заболевают сердечной проводимостью, когда электрическая система, координирующая сердцебиение, работает неправильно.
 Обратите внимание на отсутствие радиуса и большого пальца (A). Связанный с сердечно-сосудистой патологией – дефект межпредсердной перегородки. Радиографическое исследование (B и C) демонстрирует отсутствие тени радиуса; отсутствующий палец.
Обратите внимание на отсутствие радиуса и большого пальца (A). Связанный с сердечно-сосудистой патологией – дефект межпредсердной перегородки. Радиографическое исследование (B и C) демонстрирует отсутствие тени радиуса; отсутствующий палец.
Болезнь сердечной проводимости может привести к таким проблемам, как более медленный, чем ожидалось, сердечный ритм (брадикардия) или быстрое, нескоординированное сокращение сердечной мышцы (фибрилляция). Проблемы со здоровьем, связанные с сердечно-сосудистой болезнью, развиваются по мере взросления человека.
Симптомы синдрома Холта-Орама аналогичны симптомам другого расстройства, называемого синдромом Дуайн-радиального луча.
Однако они вызваны генетическими изменениями различных генов.
Холт-Орам связан с широким спектром признаков и симптомов, даже среди членов одной семьи. Это называется переменная экспрессия.
Ниже перечислены симптомы, которые могут иметь люди с этой болезнью.
| Медицинские термины | Другие имена |
80% -99% случаев | |
| Аномалия ключицы | |
| Жесткие суставы | |
| Разделенная рука | Когти руки |
Признаки встречаются 30% -79% | |
| Патология пястных костей | |
| Отсутствующий большой палец | |
| Дефект межпредсердной перегородки | |
| Атриовентрикулярный блок первой степени | |
| Кифоз | Круглая спина |
| Пароксизмальная фибрилляция предсердий | |
| Сколиоз | Искривление позвоночника |
| Трехглавый большой палец | |
| Дефект межжелудочковой перегородки | |
5% -29% людей имеют симптомы | |
| Дефект аорты | |
| Аномалия плечевой кости, ребер | |
| Нарушение атриовентрикулярного канала | |
| Широкий большой палец | |
| Закругленные, наклонные плечи | |
| Синдактилия | Сращение пальцев |
| Гипопластическое, недоразвитое сердце | |
| Радиулярный синоз | |
| Спренгельная аномалия | Высокая лопатка |
У 1% -4% людей эти симптомы | |
| Аплазия грудной мышцы, локтевой кости | |
| Гипоплазия локтевой кости | |
| Ограниченное расширение предплечья | |
| Дефект межпредсердной перегородки Secundum | |
| Короткие ключицы | |
| Syndactyly | Перфорированные пальцы |
Другие признаки | |
| Дефект позвонков | |
| Аномалия кистевых костей | |
| Частичное дублирование костей большого пальца | |
| Торакальный сколиоз |
Причина
Расстройство вызвано патогенными мутациями на TBX5. Этот ген предоставляет инструкции телу для создания белков участвующих в развитии сердца, верхних конечностей до рождения. Он особенно важен в разделении развивающегося сердца на четыре камеры, контроле роста костей рук.
Когда ген функционирует неправильно, это приводит к неправильному развитию сердца и костей, вызывает симптомы синдрома Холта-Орама.

В некоторых случаях у людей с заболеванием не обнаруживаются патогенные варианты TBX5, точная причина не понята.
Наследование
Синдром холт орама наследуется аутосомно доминантым способом. Ген TBX5 имеет пару (две копии). Автосомальная доминантность означает, что иметь только одну измененную копию TBX5 достаточно, чтобы вызвать признаки и симптомы. Человек наследует одну копию гена от матери, другую от отца. Для каждого рожденного ребенка есть:
- 50% риск наследовать измененную копию, то есть у ребенка будет болезнь;
- Пятидесяти процентный шанс наследовать здоровый ген – болезни не будет.
Примерно в 85% случаев генетическое изменение гена TBX5 происходит впервые, не наследуется от родителя.
 Коэффициент хромосомы 12 log 2, отображаемый в BlueFuse Multi v2.2 (BlueGnome), показывает дублирование 48 кб. Увеличенная область 12q24.1, точки останова 114 795 705 и 114 844 082 Mbp (NCBI GRCh37) в рамках TBX5. Дублирование включает экзоны 2-9 из транскрипта TBX5-005, экзоны 3-9 из транскрипта TBX-001.
Коэффициент хромосомы 12 log 2, отображаемый в BlueFuse Multi v2.2 (BlueGnome), показывает дублирование 48 кб. Увеличенная область 12q24.1, точки останова 114 795 705 и 114 844 082 Mbp (NCBI GRCh37) в рамках TBX5. Дублирование включает экзоны 2-9 из транскрипта TBX5-005, экзоны 3-9 из транскрипта TBX-001.
В некоторых случаях человек с синдромом Холта-Орама наследует измененную копию гена у родителя, у которого есть одна копия измененного гена, но родитель не знает, что он носитель патологии. Это происходит, потому что могут быть небольшие изменения в одной или нескольких костях запястья и незначительные изменения или нет в других костях, сердце.
Помните, изменения, вызванные синдромом Холт, могут различаться даже среди членов одной семьи. Это называется переменная экспрессия, Поскольку симптомы варьируются, невозможно предсказать, как они повлияют на будущих детей.
Диагностика
Синдром холта орама подозревается, когда у человека обнаружены изменения в костях запястья и верхних конечностей. Диагноз подтверждается, если есть определенные изменения костей, семейная история дефекта межпредсердной перегородки, дефект межжелудочковой перегородки, заболевания сердечной проводимости.

Чтобы установить диагноз, врач изучает результаты исследований, включая Рентген рук, запястий, эхокардиограмму, электрокардиограмму. Окончательно подтверждается генетическим тестированием TBX5.
Лечение
В зависимости от тяжести проблем с костями и сердцем для лечения может потребоваться команда специалистов, включая педиатров, хирургов, кардиологов, ортопедов, генетиков.
Лечение проблем с костями включает коррекционную или реконструктивную хирургию, использование протезов, физическую или профессиональную терапию. Цель – помочь людям максимально использовать верхние конечности.
Эти методы лечения наиболее эффективны, если начинаются рано. Иногда изменения кости, запястье, верхних конечностях, не вызывают никаких проблем и не нуждаются в каких-либо процедурах. В этих случаях аномалии не заметны только на Рентгене (случайный вывод).
Люди с умеренными сердечными дефектами или патологией сердечно- сосудистой системы могут не нуждаться в каком-либо лечении. В других случаях болезнь сердечной проводимости лечится антиаритмическими препаратами, кардиостимулятором для поддержания регулярного сердечного ритма.
Другие аномалии сердца устраняют хирургическим путем. Конкретная хирургическая процедура зависит от местоположения, тяжести сердечного дефекта.
Люди с дефектами сердца подвергаются повышенному риску бактериальной инфекции и воспаления (эндокардит). Антибиотики назначают перед хирургическими процедурами, чтобы снизить риск заражения.
Прогноз
Долгосрочные перспективы для людей с Холт-Орамом зависит от тяжести сердечных дефектов. У некоторых людей нет проблем с сердцем, или они мягкие и требуют периодического кардиологического мониторинга. В других случаях сердечные дефекты, бывают серьезными и опасными для жизни.
Различия в костях запястья, руки, плеча, приводят к физическим ограничениям, которые влияют на повседневную жизнь. Эти врожденные дефекты влияют на социальные взаимодействия, особенно для детей, чьи руки заметно отличаются от других.
Связанные заболевания
Дифференциальная диагностика включает:
- синдром сердечного приступа типа 2;
- синдром сердечной недостаточности типа 3;
- брахидактильно-длинный палец;
- нарушения, связанные с SAL4 (Окихиро и акро-почечно-глазной синдром);
- Fanconi анемия;
- дистальный 22q11.2 синдром микроделеции;
- ассоциация VACTERL;
- эмпиопатия талидомида;
- другие воздействия внутриутробного тератогена.
Понравилась статья? Поделись с друзьями:
Источник
Синдром Холта-Орама – это очень редкая и серьезная генетическая патология, передающаяся по наследству от поколения к поколению и имеющая аутосомный тип наследования. Такое заболевание является очень необычным, так как способно поражать сразу несколько жизненно важных генов. Эти гены влияют на правильное формирование скелета, а также на функциональные особенности сердечной мышцы. В этой статье мы рассмотрим, что же представляет собой синдром Холта-Орама, а также узнаем о методах его диагностики, причинах развития и основных методах лечения. Данная информация поможет вам разобраться во всех особенностях протекания данного недуга.
Синдром Холта-Орама: что собой представляет
Данное заболевание предупредить невозможно, так как оно имеет наследственный характер. Патология опасна не только тем, что нарушает работу сердца, но также и тем, что оставляет свой отпечаток на состоянии верхних конечностей. При этом чем больше страдает сердце, тем хуже будет развиваться и скелет. Именно поэтому является очень важным распознать синдром Холта-Орама как можно раньше, для того чтобы была возможность предотвратить самые тяжелые последствия. С этой целью проводится просто огромное количество различных генетических исследований, на фоне которых и можно определить точный диагноз. Также проведения различных методов диагностики позволяет определить, будет ли возникать патология и у будущих поколений.

Каждая беременная женщина во время вынашивания плода в обязательном порядке должна проходить ультразвуковое обследование. Такая процедура поможет уже на ранних стадиях определить, будет ли иметь малыш отклонения в строении его костной ткани. Если же таковые имеются, врачи должны проверить и состояние сердца будущего малыша. Однако чаще всего определить симптомы болезни можно только после того, как ребенок родится.
Какими бывают врожденные дефекты
Синдром Холта-Орама, фото которого вы сможете посмотреть на данном ресурсе, чаще всего накладывает свой отпечаток на строение именно верхней части скелета. Обычно пациенты страдают различными аномалиями кистей или предплечий, которые могут поражать как одну сторону тела, так и две одновременно. При этом такие аномалии могут иметь самый разнообразный характер. Например, на месте одного пальца у пациента может быть сразу две или три фаланги. Бывают случаи, когда большие пальцы на руках отсутствуют полностью.
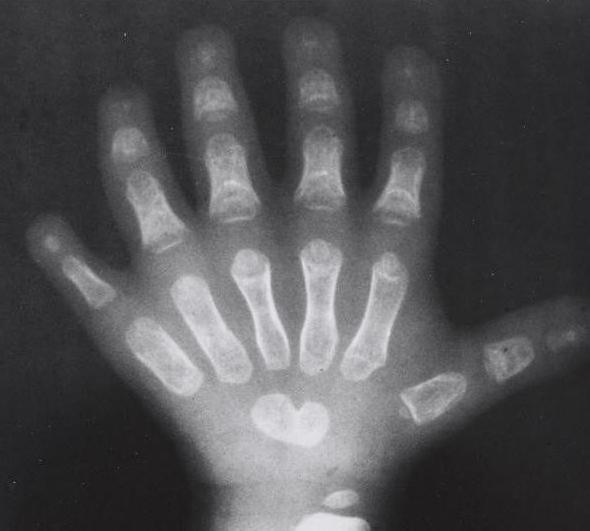
Чаще всего такое явление наблюдается именно на правой руке. Существует большое количество аномалий, касающихся такой патологии. Медицине известны случаи, когда малыши рождались без предплечий вовсе, или же их лучевые кости были недоразвитыми. Также болезнь может давать о себе знать искривлением позвоночника или неправильно сформированной грудиной, или ключицами. Чаще всего недоразвитые конечности доставляют пациентам только эстетические неприятности. Однако стоит учитывать, что заболевание также отражается и на здоровье сердечной мышцы. Практически все пациенты, страдающие синдромом Холта-Орама (синдром рука-сердце), имеют также патологии в развитии сердечной мышцы. При этом заболевания могут носить как легкий характер, так и достаточно тяжелый. Не исключены случаи развития сердечной недостаточности.
Как наследуется данная патология
Синдром Холта-Орама, описание которого вы можете прочитать в этой статье, носит доминантно-аутосомный тип наследования. Это говорит о том, что патология будет регулярно передаваться от поколения к поколению. Однако в некоторых случаях поврежденный ген не обладает высокой проявляемостью, а это говорит о том, что болезнь будет проявляться не у каждого поколения, а через одно. Чаще всего недуг передается непосредственно от родителей к детям.
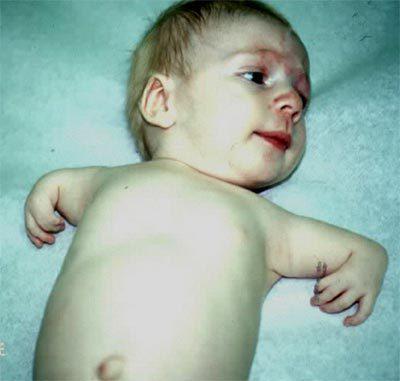
Но бывают случаи, когда ген начинает мутировать даже при условии, что у малыша родители были абсолютно здоровыми. Даже в этом случае болезнь будет передаваться следующим поколениям. Заболевание может передаваться независимо от того, кто оказался носителем мутированного гена, мужчина или женщина.
Синдром Холта-Орама: причины развития патологии
Как было сказано выше, чаще всего болезнь передается по наследству, от родителей к малышу. Обычно данное заболевание поражает двенадцатую хромосому. Однако недуг может возникнуть и при возникновении мутации данного гена даже у здоровых родителей.
Основная симптоматика
Согласно исследованиям специалистов, клинически заболевание может носить переменный характер. Но вот патологии верхних конечностей у малышей будут присутствовать в любом случае. При этом аномалии могут возникать как в одной части человеческого тела, так и сразу в двух. Они могут быть асимметричными или же симметричными. Аномалии в строении скелета будут присутствовать у каждого пациента, страдающего синдромом Холта-Орама (лечение заболевания будет описано ниже). У некоторых пациентов такие аномалии можно заметить только при проведении рентгенологического исследования.

Также большое количество пациентов будут иметь и заболевания сердца. Чаще всего пациенты страдают недугами, связанными с межпредсердной перегородкой или с межжелудочковой перегородкой.
Дефекты сердечной мышцы
Если при возникновении этого недуга страдает межпредсердная перегородка, то очень часто это может приводить к возникновению порока сердца, а также к другим заболеваниям, которые возникают внутри столь жизненно важного органа. Такая патология возникает в том случае, когда канал связи между левым и правым предсердием остается полностью открытым. Порок сердца врожденного характера приводит к возникновению сильной отдышки, повышенной утомляемости, а также к снижению защитных сил организма. Синдром Холта-Орама примерно в пятнадцати случаев сопровождается пороком сердца.
Проведение диагностики
Диагностические критерии синдрома Холта-Орама позволяют точно определить разновидность патологии. Для того чтобы точно определить масштабы заболевания, каждый пациент должен сделать рентген костей рук, а также грудной клетки. С помощью такого метода обследования можно увидеть патологии легких, а также развитие сердечной недостаточности.
С помощью эхокардиографии специалисты смогут установить наличие различных аномалий сердечной мышцы. Также очень важно сделать электрокардиограмму для того, чтобы определить состояние проводящей системы.
Ну и, конечно же, очень важно пройти генетическое обследование, с помощью которого можно обнаружить мутацию определенных генов.
Основные методы лечения
Обычно консервативное лечение с помощью применения различных медикаментозных препаратов может проводиться как в домашних условиях, так и в стационаре. Также очень часто проводится хирургическое вмешательство, целью которого является улучшить внешний вид конечностей и подправить здоровье сердечно-сосудистой системы. На сегодняшний день большинство заболеваний сердца, которые возникают при наличии такого недуга, как синдром Холта-Орама, можно устранить при помощи проведения оперативного вмешательства.
Могут ли возникать осложнения
На самом деле очень часто данный недуг приводит к большому количеству осложнений со стороны сердечно-сосудистой системы и опорно-двигательного аппарата. Нередко на фоне синдрома Холта-Орама пациенты страдают сердечной недостаточностью, аритмией, блокадой сердца и другими заболеваниями, которые могут привести и к летальному исходу. Обычно врачи дают хорошие прогнозы на выздоровление, однако они будут зависеть, в первую очередь, от того, насколько тяжелыми являются сердечные пороки.
Диагностика врожденного порока сердца
Если у новорожденного малыша существуют подозрения на развитие столь серьезного заболевания, как порок сердца, то очень важно своевременно вызвать кардиолога, для того чтобы максимально точно поставить диагноз и выбрать методы лечения данного заболевания.

В этом случае будет проведен целый комплекс диагностических мер, позволяющих определить само заболевание, а также причины его развития. Синдром Холта-Орама, патогенез которого описан в нашем обзоре, довольно часто сопровождается врожденным пороком сердца. При этом во внутриутробном периоде такую патологию удается заметить далеко не всегда.
Основные симптомы порока сердца
Чаще всего понять, что малыш родится с пороком сердца, несложно. При наличии такой патологии цвет кожи ребенка, а также его губы и уши имеют синеватый оттенок. Иногда ребенок, наоборот, становится слишком бледным, а его нижние конечности очень холодные. Если болезнь имеет врожденный характер, то обычно ее можно определить уже сразу после появления малыша на свет. Однако иногда болезнь вовсе не дает о себе знать до начала полового развития, после чего здоровье ребенка начнет ухудшаться, и все процессы развития будут протекать гораздо медленнее.
Выводы
Очень важно внимательно ознакомиться с такой патологией, как синдром Холта-Орама. Фильмы документального плана о людях, страдающих данным заболеванием, позволят понять, что это не приговор. Многие родители, думают, что их малыш, страдающий генной мутацией, не будет жить счастливой полноценной жизнью. Не стоит забывать о том, что медицина не стоит на месте. С помощью проведения различных типов хирургического вмешательства можно эстетически улучшить состояние конечностей малыша, а также увеличить их функциональность. Для этого используются специальные протезы и другие приспособления.

Также не стоит забывать, что синдром Холта-Орама в большинстве случаев сопровождается наличием внутренних заболеваний сердца. Поэтому при наличии дефектов скелета еще во внутриутробном периоде очень важно проверить также сердце малыша. Некоторые заболевание незначительные, поэтому не требуют кардинальных методов лечения. Другие же являются достаточно опасными, поэтому нуждаются в проведении хирургического вмешательства. В любом случае врачи дают очень хорошие шансы на то, что пациенты, страдающие синдромом Холта-Орама, смогут иметь достаточно хороший уровень жизни. Для этого очень важно вовремя выявить патологию и начать своевременно ее лечить.
Начните заботиться о своем здоровье, а также о здоровье ваших детей прямо сейчас. И тогда вам не будут страшны никакие заболевания. Будьте здоровы, любите себя и всегда верьте в то, что жизнь прекрасна. И она принесет вам много счастья и радости.
Источник