Синдром х 4 хромосома это

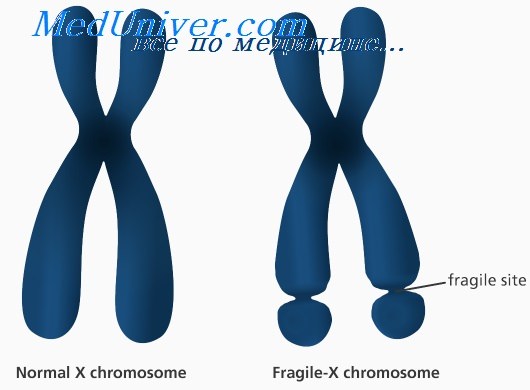
Синдром ломкой Х-хромосомы: причины, диагностика, лечениеЭтиология и встречаемость синдрома ломкой Х-хромосомы. Синдром ломкой Х-хромосомы (MIM №309550) — Х-сцепленное заболевание с задержкой умственного развития, вызванное мутациями в гене FMR1 в Xq27.3. Синдром ломкой Х-хромосомы встречается с частотой 16-25 на 100 000 в общей популяции среди мужчин и в два раза реже среди женщин. Синдром ломкой Х-хромосомы составляет 3-6% всех случаев умственной отсталости среди мальчиков с положительным семейным анамнезом по умственной отсталости при отсутствии врожденных пороков. Патогенез синдрома ломкой Х-хромосомыПродукт гена FMR1, FMRP, экспрессируется во многих типах клеток, но наиболее сильно в нейронах. FMRP может сопровождать определенный подкласс мРНК от ядра к рибосомам. Более 99% мутаций в гене FMR1 — экспансия нуклеотидного повтора (CGG)n в 5′-нетранслируемом участке гена. В нормальных аллелях FMR1 число повторов CGG составляет от 6 до приблизительно 50. В патогенных аллелях (или при полных мутациях) количество повторов более 200. Аллели с более чем 200 повторами CGG обычно имеют гиперметилированную последовательность повторов CGG и смежного промотора FMR1. Гиперметилирование инактивирует промотор FMR1, вызывая снижение экспрессии FMRP. Полные мутации возникают из аллелей премутации (от 59 до 200 повторов CGG) с передачей мутантного аллеля FMR1 от матери (но не от отца); фактически при отцовской передаче премутации часто, наоборот, сокращаются. Полные мутации не могут возникать из нормальных аллелей. Поскольку длина неустойчивых повторов CGG увеличивается в каждом последующем поколении, если они передаются женщиной, обычно наблюдается увеличение числа пораженных потомков в последующих поколениях в семье; этот феномен называется генетической антиципацией. Риск экспансии премутации в полную мутацию возрастает с увеличением числа повторов в премутации. Тем не менее не все премутации одинаково предрасположены к экспансии. Хотя премутации встречаются сравнительно часто, переход в полную мутацию наблюдают только в ограниченном количестве гаплотипов, т.е. когда есть склонность гаплотипа к экспансии. Эта склонность гаплотипа частично может быть связана с присутствием нескольких триплетов AGG, вставленных в последовательность повторов CGG; оказывается, такие триплеты AGG тормозят экспансию повторов CGG, следовательно, их отсутствие в некоторых гаплотипах может предрасполагать к экспансии.
Фенотип и развитие синдрома ломкой Х-хромосомыСиндром ломкой Х-хромосомы вызывает умеренную умственную отсталость у мужчин и легкую умственную задержку у женщин. Наиболее пораженные индивидуумы также имеют поведенческие аномалии, включая гиперактивность, размахивание руками, истерики, плохой зрительный контакт и признаки аутизма. Физические характеристики мужчин изменяются с пубертатом. До полового созревания пораженные мальчики имеют несколько увеличенный размер головы и некоторые другие неотчетливые симптомы; после наступления половой зрелости у них частые более отчетливые признаки (длинное лицо с выдающейся челюстью и лбом, крупные ушные раковины, макроорхидизм). Поскольку эти клинические признаки не уникальны для синдрома ломкой Х-хромосомы, диагноз зависит от молекулярного обнаружения мутаций. Пациенты с синдромом ломкой Х-хромосомы имеют нормальную продолжительность жизни. Почти все мужчины и 40-50% женщин, унаследовавших полную мутацию, будут иметь синдром ломкой Х-хромосомы. Тяжесть фенотипа зависит от мозаицизма метилирования повторов и их числа. Поскольку полные мутации неустойчивы, некоторые пациенты имеют смесь клеток с числом повторов, колеблющимся от премутации до полной мутации (мозаицизм числа повторов). Все мужчины с мозаицизмом числа повторов больны, но часто имеют более высокие показатели умственного развития, чем пациенты с полной мутацией в каждой клетке; у женщин с мозаицизмом числа повторов клинические проявления варьируют от нормы до полного проявления. Аналогично некоторые пациенты имеют смесь клеток с метилированием повторов CGG и без него (мозаицизм метилирования повторов). Все мужчины с мозаицизмом метилирования больны, но часто имеют более высокие показатели умственного развития, чем с гиперметилированием в каждой клетке; женщины с мозаицизмом метилирования также могут быть здоровыми или больными.
Очень редко пациенты имеют полную мутацию, неметилированную во всех клетках; независимо от пола, степень тяжести у них варьирует от нормы до полной клиники. Кроме того, у женщин фенотип зависит от степени смещения инактивации Х-хромосомы. Носительницы премутации (но не полных мутаций) имеют 20% риск ранней дисфункции яичников. Мужчины-носители премутации имеют риск развития синдрома FXTAS. FXTAS проявляет себя как поздняя прогрессирующая мозжечковая атаксия с интенционным тремором. У больных могут также присутствовать снижение краткосрочной памяти и двигательных функций, когнитивные нарушения, а также паркинсонизм, периферическая нейропатия, проксимальная мышечная слабость нижних конечности и дизавтономия. Пенетрантность FXTAS зависит от возраста, обнаруживается в 17% в течение шестого десятилетия жизни, в 38% в течение седьмого десятилетия, в 47% в течение восьмого десятилетия и в трех четвертях старше 80 лет. FXTAS может встречаться и у некоторых женщин — носительниц премутации. Особенности фенотипических проявлений синдрома ломкой Х-хромосомы: Лечение синдрома ломкой Х-хромосомыК настоящему времени никакого патогенетического лечения при синдроме ломкой Х-хромосомы нет. Помощь направлена на обучение и фармакологическое лечение поведенческих проблем. Риски наследования синдрома ломкой Х-хромосомыРиск того, что женщина с премутацией будет иметь больного ребенка, определяется размером премутации, полом плода и семейным анамнезом. Эмпирически риск для носителя перестройки иметь больного ребенка может достигать 50% для каждого мальчика и 25% для каждой девочки, но зависит от размера премутации. На основе анализа сравнительно небольшого количества матерей-носительниц известно, что риск повторения может снижаться, если премутация уменьшается со 100 до 59 повторов. Пренатальная диагностика доступна за счет использования ДНК плода из ворсин хориона или амниоцитов. Пример синдрома ломкой Х-хромосомы. Р.Л., 7-летний мальчик, направлен в клинику педиатрии в связи с умственной задержкой и гиперактивностью. Он не смог посещать детский сад, поскольку был агрессивным, не в состоянии выполнять задания, имел бедные речевые и двигательные навыки. Несмотря на задержанное развитие, он не потерял основных этапов: сидел к 10-11 мес, ходить начал в 20 мес, говорил два или три ясных слова в 24 мес. В остальном ребенок здоров. Его мать и тетя по матери имели небольшие проблемы обучения в детстве, дядя по матери умственно задержан. Данные медицинского осмотра в норме, за исключением гиперактивности. Врач рекомендовал несколько тестов, включая кариотипирование, функциональные исследования щитовидной железы и ДНК-анализ на синдром ломкой Х-хромосомы. Анализ гена FMR1 методом блот-гибридизации по Саузерну соответствовал синдрому ломкой Х-хромосомы. — Также рекомендуем «Недостаточность глюкозо-6-фосфат дегидрогеназы (Г6ФД): причины, диагностика, лечение» Оглавление темы «Генетические болезни»:
|
Источник
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 4 марта 2020;
проверки требует 1 правка.
Хромосомные болезни — наследственные заболевания, обусловленные изменением числа или структуры хромосом (геномными или хромосомными мутациями соответственно).
Хромосомные болезни возникают в результате мутаций в половых клетках одного из родителей. Из поколения в поколение передаются не более 3—5 % из них. Хромосомными нарушениями обусловлены примерно 50 % спонтанных абортов и 7 % всех мертворождений.
Все хромосомные болезни принято делить на две группы: аномалии числа хромосом и нарушения структуры хромосом.
Аномалии числа хромосом[править | править код]
Болезни, обусловленные нарушением числа хромосом в клетках человека[править | править код]
- синдром Дауна — трисомия по 21-й хромосоме (или наличие дополнительных копий генетического материала этой хромосомы по другим причинам — за счёт транслокации или дупликации);
- синдром Патау — трисомия по 13-й хромосоме, характеризуется множественными пороками развития, идиотией, часто — полидактилия, нарушения строения половых органов, глухота; большинство больных не доживают до одного года;
- синдром Эдвардса — трисомия по 18-й хромосоме, нижняя челюсть и ротовое отверстие маленькие, глазные щели узкие и короткие, ушные раковины деформированы; 60 % детей умирают в возрасте до 3 месяцев, до года доживают лишь 10 %, основной причиной служит остановка дыхания и нарушение работы сердца.
Болезни, связанные с нарушением числа половых хромосом[править | править код]
- Синдром Шерешевского — Тёрнера — отсутствие одной Х-хромосомы у женщин (45 Х0) вследствие нарушения расхождения половых хромосом; к признакам относится низкорослость, половой инфантилизм и бесплодие, различные соматические нарушения (микрогнатия, короткая шея и др.);
- полисомия по Х-хромосоме — включает трисомию (кариотип 47, XXX), тетрасомию (48, ХХХХ), пентасомию (49, ХХХХХ), отмечается незначительное снижение интеллекта, повышенная вероятность развития психозов и шизофрении с неблагоприятным типом течения;
- полисомия по Y-хромосоме — как и полисомия по X-хромосоме, включает трисомию (кариотип 47, XYY), тетрасомию (48, ХYYY), пентасомию (49, ХYYYY), клинические проявления также схожи с полисомией X-хромосомы;
- Синдром Клайнфельтера — полисомия по X-хромосомам у мальчиков (47, XXY), признаки: евнухоидный тип сложения, гинекомастия, слабый рост волос на лице, в подмышечных впадинах и на лобке, половой инфантилизм, бесплодие; умственное развитие отстает, однако иногда интеллект нормальный.
Болезни, причиной которых является полиплоидия[править | править код]
- триплоидии, тетраплоидии и т. д.; причина — нарушение процесса мейоза вследствие мутации, в результате чего дочерняя половая клетка получает вместо гаплоидного (23) диплоидный (46) набор хромосом, то есть 69 хромосом (у мужчин кариотип 69, XYY, у женщин — 69, XXX); почти всегда летальны до рождения.
Нарушения структуры хромосом[править | править код]
- Транслокации — обменные перестройки между негомологичными хромосомами.
- Делеции — потери участка хромосомы. Например, синдром кошачьего крика связан с делецией короткого плеча 5-й хромосомы. Признаком его служит необычный плач детей, напоминающий мяуканье или крик кошки. Это связано с патологией гортани или голосовых связок. Наиболее типичным, помимо «кошачьего крика», является умственное и физическое недоразвитие, микроцефалия (аномально уменьшенная голова).
- Инверсии — повороты участка хромосомы на 180 градусов.
- Дупликации — удвоения участка хромосомы.
- Изохромосомия — хромосомы с повторяющимся генетическим материалом в обоих плечах.
- Возникновение кольцевых хромосом — соединение двух концевых делеций в обоих плечах хромосомы.
В настоящее время[когда?] у человека известно более 700 заболеваний, вызванных изменением числа или структуры хромосом. Около 25 % приходится на аутосомные трисомии, 46 % — на патологию половых хромосом. Структурные перестройки составляют 10,4 %. Среди хромосомных перестроек наиболее часто встречаются транслокации и делеции.
Литература[править | править код]
- Бочков Н. П. Клиническая генетика. — М.: Медицина, 1997.
- Тоцкий В. М. Генетика. — Одесса: Астропринт, 2002.
- Шевченко В. А. Генетика человека. — М. : ВЛАДОС, 2002.
См. также[править | править код]
- Наследственные заболевания
- Генные болезни
- Полигенные болезни
- Анеуплоидия
Источник
СодержаниеТрисомии — хромосомные болезниСиндром Эдвардса: 3 месяца жизниСиндром Патау: несчастливая хромосомаСиндром Дауна: солнечные детиДругие трисомииЛечение трисомий
Человеческий организм — не машина, и в нем случаются сбои на всех уровнях: от органного до молекулярного. Особенно опасны некоторые поломки в геноме. Природа обеспечила наш вид защитными механизмами: даже если зачатие плода с тяжелыми генетическими аномалиями произойдет, с высокой долей вероятности в I триместре организм избавится от него. Но существуют тяжелые геномные патологии, при которых дети рождаются живыми, и тогда только от врачей и окружающих людей зависит, как этот ребенок проживет свою, иногда очень короткую, жизнь. В Международный день людей с синдромом Дауна MedAboutMe разбирается, какие виды трисомий, кроме синдрома Дауна, существуют и какие шансы у таких детей на выживание.
Трисомии — хромосомные болезни
Хромосомы у людей бывают двух типов: половые хромосомы, которые различаются у мужчин (XY) и женщин (XX), и аутосомы — парные хромосомы, имеющиеся у обоих полов. Для правильного развития человека, начиная с момента зачатия, важно все: и правильность строения хромосом, и их количество. Если нарушено строение, говорят о хромосомных мутациях, а если количество не соответствует норме — то о мутациях на уровне генома. Хотя отдельные хромосомные нарушения встречаются нечасто, а некоторые из них смело можно отнести к редким заболеваниям, все же в целом хромосомные аномалии фиксируются у 6-7 новорожденных из каждой тысячи.
Подавляющее большинство числовых нарушений хромосомного набора приводит к аномальному развитию плода, и организм избавляется от такого эмбриона на ранних сроках. Эксперты утверждают, что четверть спонтанных выкидышей в I триместре — это как раз результат трисомий. Но при некоторых видах анеуплоидии (утрата или, наоборот, появление дополнительной хромосомы) женщины донашивают ребенка до рождения.
Нас интересуют трисомии аутосом — ситуации, когда имеется дополнительная хромосома. Чаще всего это происходит на стадии образования яйцеклеток и сперматозоидов из-за неполного расхождения хромосом, но бывают и другие механизмы развития данной патологии. Согласно последним данным, существует даже мутация, которая существенно повышает риск передачи аномального числа хромосом потомству. В итоге, когда после оплодотворения наборы хромосом отца и матери объединяются, получившаяся клетка будет иметь трисомию — одну хромосому, как и положено, от одного родителя, и две хромосомы под тем же номером — от другого.
Трисомии могут возникать по любой из 22 аутосом человека. Но только для семи из них возможно рождение живого младенца, это 21, 18, 13, 14, 8, 9 и 22 хромосомы. В остальных случаях плод не выживает — ученые обнаруживали такие нарушения только при самопроизвольных выкидышах.
Все случаи трисомий выявляются сразу после рождения малыша. Такие дети обладают характерными чертами внешности, обычно имеют многочисленные и выраженные пороки развития, как внешних, так и внутренних органов, у них отмечается значительная задержка психомоторного развития и существенные дефекты интеллекта. Дети с большинством трисомий живут очень недолго.
Синдром Эдвардса: 3 месяца жизни
Синдром Эдвардса — результат трисомии по 18 хромосоме. Это редкое заболевание, которое встречается с частотой 1:2500-6766 среди живорожденных детей. Девочки с синдромом Эдвардса рождаются в 3 раза чаще, чем мальчики. Также доказано, что с возрастом матери риск рождения ребенка с этим заболеванием растет, впрочем, не так сильно, как в случае синдрома Дауна. Вероятность родить малыша с синдромом Эдвардса у женщин старше 45 лет составляет 0,7%.
В 70% случаев трисомии по 18 хромосоме происходит самопроизвольный выкидыш еще в I триместре беременности. Половина живорожденных детей погибает в течение первой недели жизни. Лишь 5% доживают до своего первого дня рождения. Но при этом у них обычно наличествуют множественные тяжелые пороки развития, включая скелетные и черепно-лицевые аномалии, разнообразные патологии сердца и магистральных сосудов, нарушения развития пищевода, мочевыводящей системы, желудочно-кишечного тракта. Живут такие дети в среднем не более 3 месяцев, некоторые доживают до года.
Синдром Патау: несчастливая хромосома
Синдром Патау развивается при трисомии по 13 хромосоме, которая встречается в 1 случае на 7-14 тысяч новорожденных младенцев. Мальчики и девочки с синдромом Патау рождаются с одинаковой частотой. Существует связь между риском зачатия малыша с трисомией по 13 хромосоме и возрастом матери.
Младенцы, которым удалось дожить до появления на свет, имеют многочисленные тяжелые пороки развития. Это патологии центральной нервной системы вкупе с микроцефалией, болезни глаз, деформация и недоразвитие лицевых отделов, в 80% случаев — тяжелые пороки развития сердца и сосудов, полидактилия, болезни поджелудочной железы и селезенки, почек и половых органов. У детей отмечается задержка умственного развития и глубокая идиотия. Раньше подавляющее большинство детей умирало еще до 1 года. Но сегодня, по мере развития медицины, продолжительность жизни малышей с синдромом Патау растет. В развитых странах в наше время до 5 лет доживает уже 15% детей, а до 10 лет — от 2 до 3%.
Синдром Дауна: солнечные дети
Синдром Дауна развивается при полной трисомии по 21 хромосоме. Мальчики и девочки с этой патологией рождаются с одинаковой частотой, а в среднем на свет появляется 1 ребенок с синдромом Дауна на 700 живорожденных детей. Достоверно известно, что на вероятность зачатия ребенка с такой трисомией значительное влияние оказывает возраст матери. Чем старше женщина, тем выше риски.
Как и всех обладателей трисомий, детей с синдромом Дауна отличают характерные внешние черты, по которым «солнечного» малыша можно узнать, вне зависимости от его национальности. Как и в случае других хромосомных болезней из этой группы, такие дети имеют множество патологий, нарушения умственного развития и определенных физиологических особенностей. Но, в отличие от других анеуплоидий, эта патология может развиваться без тяжелых пороков внутренних органов. И тогда такие дети имеют все шансы прожить достаточно долгую жизнь — до 60-65 лет, хоть и демонстрируя признаки раннего старения. Такой срок, сравнимый с продолжительностью жизни здорового человек — победа современной медицины, науки и, конечно, показатель развития общества, ведь еще 30-35 лет назад средняя продолжительность жизни человека с синдромом Дауна не превышала 25 лет.
Вырастая, люди с синдромом Дауна способны и сами заводить детей. Большинство мужчин бесплодны, но не все, а среди женщин могут иметь детей примерно половина. И каждый второй ребенок, рожденный матерью с трисомией по 21-й хромосоме, будет здоровым.
Другие трисомии
Другие трисомии, при которых дети имеют шанс родиться живыми, не столь известны, как вышеперечисленные три синдрома:
Трисомия по 8 хромосоме: встречается в 1 случае на 50 тысяч успешных родов. Среди проявлений патологии — макроцефалия, аномалии скелета, врожденные пороки развития мочевой системы, пороки сердца и сосудов, задержка речевого и психомоторного развития. Известны случаи, когда люди с такой патологией доживали до 17 лет. Трисомия по 9 хромосоме: микроцефалия, тяжелые нарушения опорно-двигательного аппарата, патологии сердца и сосудов, почек, желудочно-кишечного тракта. Большинство таких детей погибает в возрасте до 4 месяцев. Трисомия по 14 хромосоме: микроцефалия, пороки сердечно-сосудистой системы, тяжелые патологии почек, астма и заболевания кожи. Хотя обычно такие дети умирают достаточно рано, известны случаи, когда люди с трисомией по 14 хромосоме доживали до 13 лет. Трисомия по 22 хромосоме: рождение детей с такой патологией — большая редкость. По частоте выкидышей в I триместре эта анеуплоидия стоит на втором месте (после трисомии по 16 хромосоме). Смерть ребенка наступает обычно после рождения или в течение ближайших недель. Лечение трисомий
Пока генетические заболевания такого плана не лечатся. Однако ученые уже говорят о потенциальной возможности лечить трисомии путем генной инженерии. Например, можно было бы использовать аденовирус, как транспорт для доставки в конкретный участок лишней хромосомы гена, способного привести к ее утрате (и такой ген уже известен, по крайней мере, для 21 хромосомы). В другом варианте рассматривается возможность активизации точечных мутаций, включающих этот ген при его наличии.
По мнению экспертов, не так уж много времени осталось до момента, когда можно будет избавлять людей с такими синдромами хотя бы от тяжелых сопутствующих болезней. Например, людям с синдромом Дауна, страдающим о лейкемии, можно будет вводить «исправленные» стволовые клетки, которые способны к производству здоровых и не склонных к болезням клетки.
Неожиданный эффект дает лечение болезней сердца, которые очень часто развиваются у детей с трисомиями. У 40% детей с синдромом Дауна отмечаются врожденные пороки сердца. Когда-то это состояние было дополнительным фактором риска преждевременной смерти человека. Но в наше время такие дети получают операцию на сердце, а вместе с ней возможность жить и в значительном числе случаев быть полноценными членами общества.
В прошлом году американские ученые выступили с заявлением о необходимости проведения операций на сердце также детям с трисомиями по 13 и 18 хромосомам, то есть с синдромами Патау и Эдвардса. Из-за того, что продолжительность жизни таких малышей невелика, обычно они получают лишь симптоматическую поддерживающую терапию. Считается, что не имеет смысла делать серьезную операцию на сердце, если в течение нескольких месяцев ребенок все равно умрет. Однако врачи собрали статистику по тем детям, которые все же такую операцию получили. Оказалось, что при этом срок их жизни увеличивается на 33-67% — дети стали доживать до 2-х лет и более. Особенно выраженным эффект оказался для синдрома Эдвардса. Исследователи заявили, что это повод пересмотреть принципы проведения операций на сердце детям с трисомиями — ведь для многих родителей это шанс провести со своим ребенком не 2 недели, а 2 года.
Источник