Синдром грима кабуки рост взрослого


Синдром Кабуки – это редко встречающееся моногенное заболевание, характеризующееся своеобразными фенотипическими признаками и интеллектуальной дефицитарностью. Больные имеют лицевые особенности (миндалевидный разрез глаз, страбизм, арочные брови, широкую переносицу, низко посаженные оттопыренные уши и др.), скелетные аномалии, пороки внутренних органов, олигофрению. Диагностика основывается на клинических критериях, подтвержденных генетическими анализами. Лечение сводится к коррекции врожденных аномалий, угрожающих жизни и здоровью ребенка, терапии сопутствующих расстройств (аутоиммунных, эндокринных, инфекционных и др.).
Общие сведения
Синдром маски Кабуки (синдром грима Кабуки) получил свое название по внешнему виду и выражению лица больных, придающих им сходство с персонажами одноименного японского театра. Известно, что актеры театра Кабуки наносят на лицо специальный грим, особо выделяя глаза и брови, а в кульминационные моменты представления застывают на месте, устремив взгляд в одну точку и сводя глаза к переносице. Патология описана в 1981 году двумя авторами, представившими наблюдения за пациентами с характерными фенотипическими чертами, и в их честь именуется также синдромом Ниикава-Куроки. Синдром относится к категории орфанных: в Японии его частота составляет 1:32000, на Западе – 1:86000. На сегодняшний день описано порядка 350 случаев заболевания.

Синдром Кабуки
Причины
Патология имеет гетерогенную этиологию. Известно, что синдром является врожденным. Большая часть диагностированных случаев заболевания носит спорадический характер и развивается вследствие мутаций, возникающих «де ново». Факторы, влияющие на изменчивость, не определены. В 2010-2012 г.г. изучены две наиболее часто регистрируемые наследственные формы синдрома, в основе которых лежат мутации генов:
- KMT2D (MLL2). Ген кодирует лизин-специфическую метилтрансферазу 2D, расположен в локусе 12q13.12. Мутация вызывает первый тип заболевания (KS1) с аутосомно-доминантным наследованием. Обнаруживается у 70% пациентов.
- KDM6A. Ген расположен на участке Xp11.3, кодирует лизин-специфичную 6А деметилазу, выступает кофактором KMT2. Генный дефект связан с развитием второго типа синдрома (KS2), имеющего наследование, сцепленное с Х-хромосомой. Составляет около 5% случаев патологии.
В генетических исследованиях встречаются данные о сочетании синдрома Кабуки с другими наследственными заболеваниями: синдромом Фрейзера, гипокалиемическим периодическим параличом, синдромом Фанкони, сахарным диабетом 1 типа. Сообщается о наличии у больных структурных перестроек хромосом, таких как кольцевая Х-хромосома, транслокации, дупликации, инверсии 8 хромосомы. Относительно небольшая выборка пациентов и сравнительно недавнее внедрение молекулярно-цитогенетических методов диагностики оставляют перспективы для дальнейших исследований этиологии синдрома.
Патогенез
Ввиду неполной изученности причин синдрома Ниикава-Куроки патологические механизмы также остаются недостаточно ясными. Наиболее освещены роль и влияние мутаций в генах KMT2D и KDM6A. Оба гена выступают эпигенетическими регуляторами модификации гистонов (ядерных белков) и играют важную роль в процессе экспрессии генов. Дефекты кодируемых ими ферментов изменяют метилирование лизина в гистоне H3, что приводит к развитию гетерогенных врожденных аномалий (черепно-лицевых, скелетных, висцеральных), неврологических и эндокринных нарушений. Кроме этого, KMT2D и KDM6A принимают участие в дифференцировке наивных CD4+ Т-лимфоцитов в иммунокомпетентные клетки. Этим объясняется тот факт, что у многих пациентов с синдромом Кабуки отмечаются иммунные нарушения, проявляющиеся как снижением противоинфекционной защиты, так и аутоиммунными расстройствами.
Симптомы
Клиническая картина характеризуется множественными дефектами, затрагивающими различные анатомические системы: лицевой скелет, опорно-двигательный аппарат, кожные покровы, сердечно-сосудистую систему, ЖКТ, мочеполовые органы, анализаторы и пр. Также выявляются нарушения неврологического, эндокринного, иммунного статуса различной степени выраженности. Наиболее патогномоничны для синдрома лицевой дисморфизм, который делает лица пациентов, похожими на маску актеров театра кабуки, низкорослость и снижение интеллекта.
Лицевые аномалии включают микроцефалию, высоко поднятые (аркообразные) брови, гипертелоризм, антимонголоидную глазную щель (опущение наружных уголков глаз относительно внутренних), эктропион нижнего века. Дополняют портрет больного с синдромом Кабуки низко расположенные торчащие и деформированные уши, широкий приплюснутый нос, маленький рот с тонкими губами. Реже отмечается голубоватая окраска склер, небные расщелины, заячья губа, микрогнатия, аномалии зубов (гиподонтия, микродонтия, неправильный прикус). Своеобразное выражение лица заметно уже в раннем детстве, с годами же черты приобретают еще более грубый и гротескный вид.
Отклонения в развитии костно-суставной системы представлены деформацией позвонков, сколиозом, недоразвитием трубчатых костей, полиморфными аномалиями пальцев рук (брахидактилией, синдактилией, арахнодактилией, клинодактилией), деформацией стопы. Уже на первом году жизни обнаруживается задержка роста, в последующем низкорослость сохраняется и становится более заметной. Характерна гипермобильность суставов, являющаяся причиной частых вывихов. Со стороны кожных покровов встречается гиперэластичность кожи, наличие гипо- и гиперпигментированных пятен, гиперемия кожи лица, алопеция и гирсутизм. Важный дерматоглифический маркер – выступающие подушечки пальцев рук.
Из висцеральных пороков при синдроме Кабуки наиболее часты и клинически значимы ВПС (дефекты перегородок, единственный желудочек, коарктация аорты, тетрада Фалло, транспозиция магистральных сосудов), аномалии ЖКТ (незавершенный поворот кишечника, атрезия ануса, врожденные диафрагмальные грыжи), пороки развития мочевого тракта (гидронефроз, подковообразная почка). У девочек может отмечаться преждевременное телархе, у мальчиков – крипторхизм, гидроцеле и микропенис.
Анатомическим дефектам сопутствуют функциональные расстройства ‒ синдромы мальабсорбции и холестаза, сердечные блокады, мочеточниковый рефлюкс. Патология зрительного анализатора включает миопию, астигматизм, катаракту, птоз, косоглазие. У большинства детей диагностируется тугоухость.
У 80% пациентов обнаруживаются неврологические нарушения: гипотонус мышц, тремор, судороги, эпилепсия, ЗПРР. Степень олигофрении варьирует от слабой до умеренно выраженной. Могут наблюдаться черты аутистического поведения, обсессивно-компульсивный синдром.
Ввиду дефектов иммунной регуляции и снижения уровня сывороточных Ig отмечаются частые инфекции, аутоиммунные патологии (тромбоцитопеническая пурпура, геморрагический васкулит.), гематологические заболевания (гемолитическая анемия). Из гормональных нарушений наиболее часты СД 1 типа, врожденный гипотиреоз, снижение секреции СТГ и АКТГ, ожирение.
Осложнения
Потенциальные осложнения тесно связаны с теми мультисистемными поражениям, которые имеются у ребенка. У новорожденных возможны серьезные нарушения сосания и глотания, требующие установки желудочного зонда или гастростомы. Также в младенчестве отмечаются необъяснимые эпизоды гипогликемии, угрожающие переходом в гипогликемическую кому. Врожденные пороки сердца могут вызывать серьезные гемодинамические расстройства, требующие проведения кардиохирургических вмешательств. Частые ОРВИ нередко осложняются отитами, пневмониями. Прогрессирующая атрофия зрительных нервов может приводить к слепоте. У больных с синдромом Кабуки имеется повышенный риск развития лимфолейкоза и неходжкинских лимфом.
Диагностика
Основная проблема диагностики – низкая информированность специалистов первичного звена о данном синдроме, отсутствие четко выработанных клинических критериев и методов лабораторной верификации заболевания. Диагноз в основном устанавливается на основании фенотипичеких маркеров; в крупных медицинских центрах доступно генетическое тестирование. Первичная диагностика проводится генетиком, однако пациенты с синдромом Кабуки требуют обследования и курации целым рядом специалистов: педиатром, кардиологом, неврологом, ортопедом, урологом, иммунологом, ортодонтом и т. д. Этапы обследования включают:
- Портретную диагностику. Ядро признаков составляют лицевые стигмы, отставание в росте, скелетные аномалии. Кроме высокочастотных проявлений учитываются и редко встречающиеся атипичные признаки, известные по описаниям отдельных клинических наблюдений (витилиго, множественные гемангиомы, гипоспадия).
- Генетические исследования. Выполняются в специализированных лабораторных центрах. Позволяют изучить мутационный статус генов MLL2 и KDM6A. Для проведения высокоточной диагностики используются методы флюоресцентной (FISH) и сравнительной геномной гибридизации на микрочипах (aCGH). Обязательно обследование родителей на наличие аналогичных мутаций.
- Лабораторную диагностику. Гемограмма характеризуется тромбоцитопенией, лейкопенией, нейтропенией. Для обнаружения сопутствующих нарушений проводятся гормональные и иммунологические анализы. Исследуется гликемический профиль, уровень гормонов щитовидной железы и гипофиза, иммуноглобулинов, ЦИК.
- Инструментальное обследование. Осуществляется с целью выявления сопутствующих органных нарушений. Первоочередная диагностика включает ЭхоКГ, УЗИ органов брюшной полости и почек, рентгенографию костей и позвоночника. При эписиндроме выполняется видео-ЭЭГ-мониторинг. Дополнительно проводится офтальмологическое обследование, аудиометрия.
При высоких рисках рождения ребенка с генетическими аномалиями назначается инвазивная пренатальная диагностика с последующим полногеномным секвенированием. Дифференциальная диагностика осуществляется с другими генетическими синдромами, сопровождающимися множественными нарушениями развития: Ди Джорджи, Ван-дер-Вуда, Мельника-Фрейзера, Элерса-Данло, Чарджа и другими.
Лечение синдрома Кабуки
Ввиду генетической детерминированности заболевания полное излечение невозможно. Стандарты терапии синдрома не регламентированы, на практике применяется симптоматический подход к устранению тех нарушений, которые мешают развитию ребенка. Хирургическая патология (грыжи, ВПС, расщелины ЧЛО, атрезия анального канала, крипторхизм и т.п.) устраняются в ходе оперативного лечения.
Сопутствующие аутоиммунные расстройства корригируются назначением глюкокортикоидов, иммуносупрессоров, моноклональных антител. Рецидивирующие инфекционные процессы диктуют необходимость применения антибактериальных средств, внутривенного введения человеческого иммуноглобулина. При выявлении диабета 1-го типа решается вопрос о назначении инсулинотерапии.
Прогноз и профилактика
Дети, страдающие синдромом Кабуки, нуждаются в постоянном уходе и наблюдении, социальной адаптации, инклюзивном образовании. Семье, воспитывающей такого ребенка, требуется психологическая поддержка. В связи с большим количеством сложных дефектов (анатомических, иммунологических, эндокринных, неврологических) качество и продолжительность жизни пациентов снижены. Профилактика спорадических случаев синдрома не разработана, ведущую роль здесь отводится грамотной прегравидарной подготовке, исключению воздействия мутагенных факторов среды. Выяснить вероятность наследования синдрома Кабуки ребенком при наличии семейных случаев патологии позволяет генетическое консультирование.
Источник
Синдром Кабуки — это редкое генетическое заболевание, встречающееся раз на 32000 новорожденных детей. Внешний вид больного ребенка (фенотип) напоминает грим актера древнейшего японского театра Кабуки.
Перед выступлением проводилось длительное гримирование мужчин-актеров. Внешние углы глаз натягивали вверх и в сторону. В некоторых источниках используется термин «синдром грима Кабуки». Иногда встречается эпонимическое название — синдром Ниикава-Куроки. Глаза особенно хотели выделить. Многие актеры жертвовали своим здоровьем и красили глазные яблоки. Также выделяли брови, делая их изогнутыми дугой. Когда представление заканчивалось, персонаж замирал на месте, устремлял свой взгляд в одну точку и косил глаза.

Заболевание проявляется характерными чертами лица в совокупности с умственной отсталостью. Изучая семьи с синдромом Кабуки, выявили аутосомно-доминантный тип наследования.
Историческая справка
Описан в 1981 году японскими врачами Ниикава и Куроки. Первое исследование показало, что из 60 детей 58 были японцами. Больше десятилетия в медицинском мире считали синдром японским недугом. В 1992 году стало очевидным, что заболеванию подвержены люди любой расовой принадлежности. Случаи синдрома были выявлены в Северной Америке, арабских странах, Беларуси. По настоящее время было изучено более 350 случаев патологии.

Причины синдрома
Этиология до конца не изучена. Проводилось множество исследований. Изучали отношение половой принадлежности к заболеванию (50/50), исключили связь с близкородственными браками, не выявили патологических влияний на плод во время беременности. Проводили кариотипирование всех больных детей и их родителей. Находили генные мутации и пытались их подвести под синдром, но исследования опровергались.
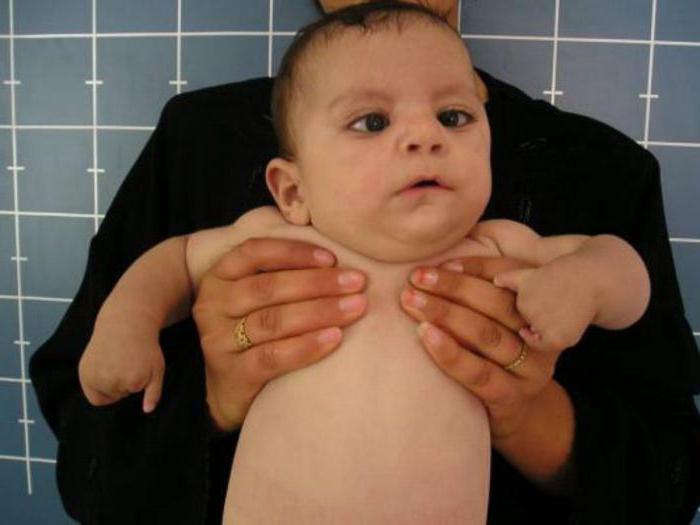
В 2011 году проводился скрининг в 110 семьях с синдромом Кабуки, и в 81 случае была обнаружена мутация гена MLL2, но это не абсолютный показатель заболевания. Диагноз до сих пор ставится на основании клинической картины. Исследования ведутся до сих пор.
Клинические признаки заболевания
На основании большого количества обследованных ученый Ниикава с соавторами выделил 5 кардинальных симптомов синдрома Кабуки.
- Характерное лицо — длинные глазные щели, густые ресницы, выворот нижнего века (эктропион), широкая переносица, приплюснутый кончик носа, большие оттопыренные уши, брови в виде арок с редким ростом в их боковой части, низкий рост волос.
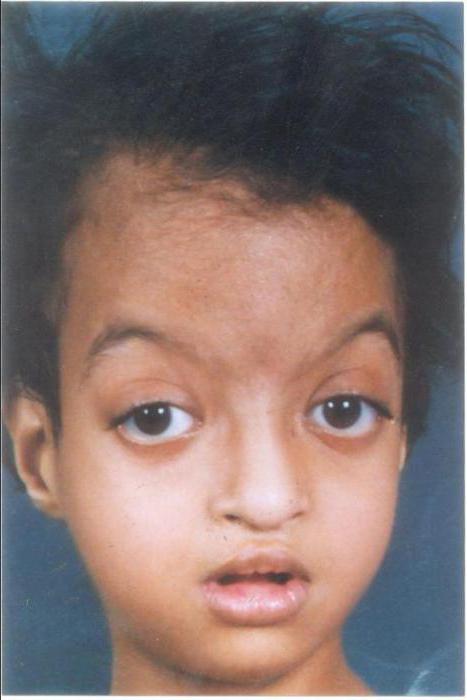
- Скелетные аномалии — патология черепа и микроцефалия, готическое небо (высокое), расщелина твердого неба в сочетании с заячьей губой, зубные аномалии (широкие зубные промежутки, недостаточно зубов, формируется неправильный прикус), задержка роста, короткие пальцы рук (особенно 5-го), крестцово-копчиковая пазуха (эпителиально-копчиковый ход), повышенная подвижность суставов, сколиоз.
- Изменение дерматоглифики — фетальные подушечки на кончиках пальцев.
- Интеллектуальный дефицит различной степени. Тест IQ в среднем 60-80 баллов. Дети с синдромом Кабуки чаще подвержены аутизму. Характерно нарушение поведения, бедность эмоций, тенденция к жеванию несъедобных предметов.
- Постнатальная задержка роста.
Другие симптомы
Они сопровождают основные симптомы болезни. Поражаться может любая система организма.
- Патология сердечно-сосудистой системы. Может сформироваться любой врожденный порок сердца (тетрада Фалло, дефект межжелудочковой или межпредсердной перегородки, коарктация или аневризма аорты, открытый артериальный проток).
- Хирургическая патология — грыжи (пупочная, паховая, диафрагмальная), атрезия ануса.
- Сбои в пищеварительной системе в виде нарушения всасывания питательных веществ (мальабсорбция), дискинезия желчевыводящих путей.
- Патология мочевыводящей системы — дисплазия, подковообразная почка, расщепление почечной лоханки, удвоение и обструкция мочеточников.
- Пороки развития наружных половых органов — крипторхизм, микропенис.
- Иммунодефицит. У детей часто возникают инфекции верхних дыхательных путей и пневмонии. Наблюдается обострение средних отитов.
- Эндокринная патология. В младенческом возрасте может быть гипогликемия. Иногда развивается сахарный диабет, врожденный гипотиреоз, ожирение. Уровень соматотропного гормона может быть снижен. У девочек может быть преждевременное половое развитие.
- Гематологические нарушения — идиопатическая тромбоцитопеническая пурпура, аутоиммунная гемолитическая анемия, полицитемия, неонатальная гипербилирубинемия.
- Нарушения со стороны органов чувств. Нарушение зрения (миопия, астигматизм, неполная атрофия зрительного нерва. Снижение или потеря слуха (65% случаев).
- Неврологическая патология. С самого рождения может быть снижение сосательного и глотательного рефлексов. Паралич отводящего нерва, нистагм, косоглазие, опущение века. У трети детей наблюдается мышечная гипотония, которая может длительно сохраняться. С взрослением дети отстают в психологическом и физическом развитии, наблюдается нарушение координации движений, задержка речевого развития. Может быть тремор кистей и стоп. Развитие эпилепсии встречается в любой возрастной категории, больше подвержены девочки.
Диагностика синдрома Кабуки
Диагноз ставится на основании клинической картины и подтверждается хромосомными исследованиями. Проводится молекулярное исследование на предмет мутации гена MLL2. Некоторые генетические заболевания имеют схожие черты, необходимо провести дифференциальную диагностику.
Важно выявить серьезные нарушения органов и систем с помощью широкого спектра лабораторно-инструментальных исследований.
Лечение синдрома Кабуки
Специфической терапии не существует. Проводится коррекция имеющихся нарушений в организме. Проводится хирургическое лечение пороков сердца. Осуществляют пластику твердого неба. При нарушении слуха необходимо лечение у отоларинголога и сурдолога, решается вопрос о слуховом аппарате. Компенсируются другие нарушения. Дети с синдромом Кабуки нуждаются в адаптации к социальной среде. Семьям требуется психологическая и материальная поддержка для воспитания такого ребенка.
В январском номере ведущего американского журнала PNAS была опубликована статья о лечении умственной отсталости при синдроме Кабуки. Суть терапии заключается в применении низкоуглеводной кетоновой диеты. Такое питание помогает добиться повышения уровня нейрогенеза. Исследование проводилось на лабораторных мышах в университете Джонса Хопкинса. Доказано было повышение уровня восстановления нервной ткани в мозгу животных.
Наша соотечественница, доктор биологических наук Дьяконова Варвара прокомментировала открытие американских коллег. Она считает, что пищевая коррекция значительно безопаснее химических лекарств. Имеются предпосылки, что данный способ лечения поможет в лечении других генетических аномалий, сопровождающихся умственной отсталостью.

Известно, что кетоновые тела — это метаболиты распада жира. Их образование происходит при недостатке в рационе углеводов и переходе на белковое питание. Кетоны обеспечивают питание головного мозга, но в то же время их избыток оказывает отравляющее действие на центральную нервную систему. Поэтому внедрение подобной диеты в рацион больных детей требует дополнительных испытаний. Ведутся исследования.
День осведомленности о заболевании — 23 октября
Отмечается во многих странах мира. Создана специальная Межрегиональная Ассоциация детей-инвалидов и их родителей «Синдром Кабуки». Работа общественной организации заключается в поддержке семей, столкнувшихся с данной патологией. В социальных сетях существуют блоги родителей, которые делятся своим опытом в воспитании и адаптации больного ребенка. Люди рассказывают о проблеме, как с ней жить.

Заболевание и прогноз
У синдрома Кабуки прогноз благоприятный. На продолжительность жизни влияют наличие патологии со стороны сердечно-сосудистой системы и степень иммунной защиты.
Источник