Синдром горнера код мкб 10

Все материалы
Клинические протоколы МЗ РК
ҚР ДСӘДМ клиникалық хаттамалар
Клинические рекомендации МЗ РФ
Клинические протоколы МЗ РБ
Международные клинические руководства
Обзорные статьи по заболеваниям
- Ещё
Клинические протоколы МЗ РК — 2020
Клинические протоколы МЗ РК — 2019
Клинические протоколы МЗ РК — 2018
Клинические протоколы МЗ РК — 2017
ҚР ДСӘДМ клиникалық хаттамалар — 2017
Клинические протоколы МЗ РК — 2016
Клинические протоколы МЗ РК — 2015
Клинические протоколы МЗ РК — 2014
ҚР ДСӘДМ клиникалық хаттамалар — 2014
Клинические протоколы МЗ РК — 2013
Архив — Клинические протоколы МЗ РК — 2012 (Приказы №883, №165)
Архив — Клинические протоколы МЗ РК (Протокол №8 от 17.04.2012 г., Экспертный совет МЗ РК)
Архив — Клинические протоколы МЗ РК — 2010 (Приказ №239)
Архив — Клинические протоколы МЗ РК — 2007 (Приказ №764)
Архив — Аурулардың диагностикасы және емдеу хаттамалары (Приказ №764, 2007, №165, 2012)
Архив — Протоколы диагностики и лечения Министерства здравоохранения Республики Казахстан (2006, устар.)
Клинические протоколы (Беларусь)
Клинические рекомендации РФ (Россия) 2013-2017
Клинические рекомендации РФ (Россия) 2018-2020
Международные клинические руководства для использования в РК
Источник
Содержание
- Синонимы диагноза
- Описание
- Симптомы
- Причины
- Лечение
Другие названия и синонимы
Синдром марионетки, Синдром Петрушки, Синдром счастливой куклы.
Названия
Синдром Ангельмана.
Дефект в 15-й хромосоме — Причины Синдром Ангельмана
Синонимы диагноза
Синдром марионетки, Синдром Петрушки, Синдром счастливой куклы.
Описание
Cиндром Ангельмана является генетическим заболеванием, которое вызывает отклонения в развитии пациента, неврологические проблемы в виде нарушения речи, трудности при ходьбе и, в некоторых случаях, судороги.
Синдром Ангельмана обычно не обнаруживается, пока родители не начинают замечать задержку в развитии ребенка в возрасте 6-12 месяцев. Явные дефекты проявляются в возрасте 2 — 3 лет.
Как правило, синдром Ангельмана не влияет на продолжительность жизни.
Симптомы
Признаки синдрома Ангельмана включают в себя:
1. Задержка в развитии ребенка.
2. Отсутствие или минимальный объем речи.
3. Неспособность ходить, нарушение равновесия (атаксия).
4. Тремор при движении рук и ног.
5. Ребенок часто улыбается и смеется.
6. Повышенная возбудимость личности.
Люди с синдромом Ангельмана может также иметь другие признаки и симптомы, в том числе:
1. Судороги, которые обычно начинаются с 2 — 3 лет.
2. Резкость движений.
3. Уменьшенный размер головы, скошенный затылок (микробрахицефалия).
4. Косоглазие.
5. Точкообразные движения языка.
6. Гипопигментации волос, кожи и радужки глаз.
Большинство детей с синдромом Ангельмана не проявляют признаков расстройства при рождении. Первые симптомы в виде задержки в развитии проявляются в возрасте 6 — 12 месяцев.
Миоклония. Судороги. Тремор.
Причины
Синдром Ангельмана является генетическим расстройством. Чаще всего он связан с проблемами гена, расположенного в 15-ой хромосоме (ген UBE3A).
Обычно, только материнская копия гена UBE3A активна в мозге. Большинство случаев синдрома Ангельмана развивается, когда часть материнской 15-й хромосомы, содержащая этот ген, отсутствует или повреждена. В небольшом числе случаев, этот синдром развивается, когда наследуются две отцовские копии, вместо одной отцовской и одной материнской копии (отцовская дисомия).
Синдром Ангельмана встречается редко. В большинстве случаев исследователи не знают, что вызывает генетические изменения, которые приводят к синдрому Ангельмана. Большинство людей с этой патологией не имеют наследственной истории этого заболевания. В небольшом проценте случаев, однако, синдром Ангельмана может быть унаследован от родителей.
Лечение
Так как нет способов восстановления хромосомных дефектов, нет и этиотропного лечения для синдрома Ангельмана.
В таких случаях проводится симптоматическое лечение, обусловленное проявлениями синдрома. В зависимости от симптомов, лечение синдрома Ангельмана может включать в себя следующие пункты:
1. Антиконвульсанты. Лекарства могут быть необходимы для контроля припадков, вызванных синдромом Ангельмана.
2. Лечебная физкультура. Дети с синдромом Ангельмана могут научиться ходить лучше и преодолеть другие проблемы с движениями с помощью физиотерапии.
3. Хотя люди с синдромом Ангельмана обычно не овладевают вербальным языком в полной мере, общение очень полезно для таких пациентов. Невербальные языковые навыки могут быть разработаны на основе языка жестов.
4. Поведенческая терапия. Поведенческая терапия может помочь детям с синдромом Ангельмана преодолеть гиперактивность, улучшить концентрацию внимания. Многие больные с синдромом Ангельмана способны строить отношения с друзьями и создать семью.
Источник
Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Описание
- Дополнительные факты
- Симптомы
- Причины
- Течение и стадии
- Диагностика
- Лечение
Названия
Название: Синдром Драве.

Синдром Драве
Описание
Синдром Драве. Это детская энцефалопатия наследственного характера, которая характеризуется эпилептиформными приступами, отставанием в психическом развитии и резистентностью к противоэпилептической терапии. Клинически заболевание проявляется полиморфными эпилептическими припадками, неврологическими расстройствами, атипическими абсансами и фокальными моторными пароксизмами. Диагностика синдрома Драве базируется на характеристике возникающих приступов, данных ЭЭГ и МРТ, идентификации мутации генов SCN1A или GABRG2. Лечение малоэффективно и проводится с целью уменьшения частоты приступов, профилактики эпилептического статуса.
Дополнительные факты
Синдром Драве или тяжелая миоклоническая эпилепсия младенчества – это аутосомно-доминантная энцефалопатия с дебютом в первые 12 месяцев жизни ребенка, которая проявляется фебрильными и афебрильными генерализованными приступами, фокальными миоклоническими пароксизмами, расстройствами неврологического статуса и дефицитом интеллекта. Впервые заболевание было описано французским психиатром и эпилептологом Шарлоттой Драве в 1978 году. Встречается данный синдром редко, распространенность – 1:20-40 тысяч детского населения. У мальчиков патология возникает вдвое чаще, чем у девочек. Исход синдрома Драве неблагоприятный – заболевание неизлечимо и слабо поддается медикаментозной терапии. Летальность составляет порядка 16-18%.
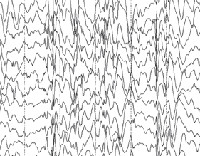
Синдром Драве
Симптомы
Клонические судороги. Миоклония. Судороги. Тремор.
Причины
Синдром Драве – это генетически детерминированная патология, которая передается по аутосомно-доминантному типу наследования. Спровоцировать развитие тяжелой миоклонической эпилепсии младенчества могут мутации локуса SCN1A на 24 участке длинного плеча 2 хромосомы (в 80% случаев) или GABRG2 на 5q34. Данные гены кодируют α1-субъединицу Na+-каналов, что приводит к нарушению физиологических процессов реполяризации и деполяризации в нейронах, и как следствие – к патологической активности ЦНС.
Течение и стадии
В клинической картине синдрома Драве выделяют 3 этапа развития: фебрильный (до 12-24 месяцев), агрессивный или катастрофический (2-8 лет), статический (старше 8 лет). Дебют заболевания происходит в возрасте от 2 месяцев до 1 года, в среднем – в 5 месяцев. До момента возникновения первых симптомов ребенок развивается нормально, неврологических и психических отклонений не наблюдается. В большинстве случаев первичными проявлениями фебрильной стадии синдрома Драве становятся фибриллярные судороги атипического характера. Они имеют большую продолжительность (свыше 20 минут), включают в себя очаговые компоненты и альтернирующие гемиконвульсии, иногда переходят в эпилептический припадок. На ранних этапах такие состояния сопровождаются субфебрильной или фебрильной температурой тела, в дальнейшем подобных проявлений не наблюдается. Зачастую при синдроме Драве приступ может быть спровоцирован гипертермией (согреванием, горячей ванной или инфекционной патологией), световыми раздражителями, резкими движениями.
Катастрофический или агрессивный период синдрома Драве характеризуется выраженными полиморфными клонико-тонико-клоническими припадками, альтернирующими гемиконвульсиями, очаговыми моторными пароксизмами, атипичными абсансами. Приступы начинаются с мышечных подергиваний по всему телу (иногда – асинхронных), переходят в кратковременную тоническую, а затем – клоническую фазы. Часто подобное состояние трансформируется в эпилептический статус, который может сохраняться до нескольких суток. В возрасте 1-2 лет у больных с синдромом Драве определяется дефицит интеллекта (олигофрения) и гиперактивность, поведенческие аномалии, нарастающие до 6-7 лет и сохраняющиеся на протяжении всей жизни. Также развиваются неврологические нарушения: мышечная гипотония, атаксия, интенционный тремор, моторная неловкость, признаки пирамидной недостаточности. В этом же возрасте у части детей возникает паттерн-сенситивность, при которой определенная одежда, обои или телевизионные передачи могут стать причиной очередного приступа.
Статическая стадия синдрома Драве характеризуется уменьшением интенсивности и частоты эпилептических припадков. Психические и неврологические отклонения остаются. Большая часть приступов возникает в ночное время или сразу после пробуждения. Как и в других периодах, они могут быть спровоцированы повышением температуры тела, ярким светом, резким движением и тд На фоне отставания в интеллектуальном развитии, нарушений психики и резистентности заболевания к лечению пациент почти полностью лишен способности адаптироваться в социуме.
Диагностика
Диагностика синдрома Драве основывается на анамнестических данных, физикальном обследовании, лабораторных и инструментальных методах исследования. Из анамнеза педиатром выясняется возраст, в котором произошла манифестация патологии, первичные проявления, характеристика приступов, степень их тяжести и динамика развития. При осмотре ребенка в межприступный период можно выявить отставание в интеллектуальном развитии (ЗПР), гиперактивность, нарушения неврологического статуса. Во время припадка определяются атипичные абсансы, очаговые расстройства, альтернирующие гемиконвульсии.
Общие лабораторные анализы (ОАК, ОАМ, анализ кала) малоинформативны – выраженные отклонения от возрастной нормы, как правило, отсутствуют. Из инструментальных методов исследования при синдроме Драве используются электроэнцефалограмма (ЭЭГ) и магнитно-резонансная томография (МРТ). Между приступами на ЭЭГ у большинства таких детей определяется сочетание очаговой, мультирегиональной и диффузной эпилептиформной активности с нарастанием во сне. При низкой частоте припадков данные признаки могут отсутствовать. По результатам МРТ головного мозга удается установить признаки диффузной атрофии коры головного мозга и мозжечка, субкортикальных слоев, иногда – увеличение размеров желудочков. Для подтверждения синдрома Драве используется кариотипирование с определением мутации генов SCN1A или GABRG2.
В педиатрии дифференциальная диагностика синдрома Драве проводится с фебрильными судорогами, митохондриальными и дисметаболическими патологиями, доброкачественной миоклонической эпилепсией младенчества, синдромами Леннокса-Гасто и Дозе, другими формами эпилепсии у детей, которые сопровождаются миоклоническими припадками. Практически идентичную клиническую картину имеет мутация гена PCDH19 – эпилепсия с умственной отсталостью, ограниченная женским полом.
Лечение
Синдром Драве – это форма эпилепсии у детей, которая почти не поддается терапии. Основная цель лечения – снизить чистоту приступов, профилактировать их трансформацию в эпилептический статус. Как правило, большинство распространенных противоэпилептических средств при тяжелой миоклонической эпилепсии младенчества неэффективны. В качестве стартовой терапии показаны вальпроаты (вальпроева кислота) и сульфат-замещенные моносахариды (топирамат). Также могут применяться фармакологические средства из групп барбитуратов и бензодиазепинов. В некоторых случаях при синдроме Драве позитивная динамика отмечается на фоне кетогенной диеты, которая подразумевает большое количество жиров и строгое ограничение углеводов.
Прогноз для жизни при синдроме Драве сомнительный, для выздоровления – неблагоприятный. Дефицит интеллекта, расстройства психики, эпилептические припадки и неврологические нарушения обычно сохраняются на протяжении всей жизни человека, что обусловливает его полную социальную дезадаптацию. Обычно приступы возникают в ночное время или сразу после пробуждения, а их интенсивность и частота уменьшаются. Смертность составляет порядка 15,9-18%. Основные причины – синдром внезапной детской смерти при эпилепсии, интеркуррентные инфекционные заболевания, несчастные случаи во время припадков.
Антенатальная профилактика синдрома Драве аналогична другим наследственным заболеваниям. Она подразумевает медико-генетическое консультирование и планирование беременности, кариотипирование плода посредством амнио- или кордоцентеза. Постнатальные превентивные меры включают в себя исключение гипертермических состояний у ребенка (раннее лечение инфекционных заболеваний, избегание горячих ванн ) и других факторов, которые могут спровоцировать приступ.
Источник
Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Содержание
- Описание
- Симптомы
- Причины
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Синдром Рейно.
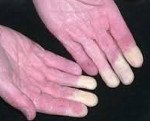
Синдром Рейно
Описание
Синдром Рейно — пароксизмальное вазоспастическое расстройство артериального кровоснабжения кистей и (или) стоп, возникающее чаще под воздействием холода или волнений.
Симптомы
• Поражаются преимущественно II-V пальцы кистей стоп, реже нос, уши, подбородок.
• Во время приступа — парестезии, кожа мертвенно-бледная, пальцы холодные.
• По окончании приступа — болезненность, чувство жара и распирания, кожа гиперемирована.
• Постепенно развиваются трофические изменения (уплощение или втяжение подушечек пальцев, снижение тургора кожи, плохо заживающие язвы).
• В межприступный период кисти холодные, цианотичные, влажные.
• Возможно наличие внутренних эквивалентов в сосудах сердца, легких, почек, мозга, с поражением этих органов, особенно при системной склеродермии.
Диагностические критерии:
• Эпизоды двусторонних изменений цвета кожи, обусловленные холодом или эмоциями.
• Сохраненная пульсация на периферических артериях.
• Симметричность поражения.
• Отсутствие гангрены.
• Отсутствие признаков системного заболевания.
• Продолжительность заболевания не менее 2 лет.
Озноб.
Синдром Рейно
Причины
Развивается при ряде заболеваний:
• диффузные заболевания соединительной ткани, в первую очередь системная склеродермия;
• ревматоидный артрит;
• системные заболевания сосудов (артериальная гипертензия, васкулиты, узелковый периартериит);
• заболевания нервной системы (симпатических нервных узлов);
• болезни крови, парапротеинемия, полицитемия, криоглобулинемия;
• эндокринные заболевания (токсический зоб);
• диэнцефальные расстройства;
• регионарная компрессия сосудисто-нервного пучка (добавочные шейные ребра, поражение шейного отдела позвоночника, синдром передней лестничной мышцы);
• воздействие профессиональных факторов (вибрация, охлаждение).
Лечение
Лечение синдрома Рейно — сложная задача, решение которой зависит от возможности устранения причинных факторов и эффективного воздействия на ведущие механизмы развития сосудистых нарушений.
Всем больным с синдромом Рейно рекомендуется исключить охлаждение, курение, контакт с химическими и другими факторами, провоцирующими сосудистый спазм в быту и на производстве. Иногда достаточно изменить условия труда (исключить вибрацию и ) или место жительства (более теплый климат), чтобы проявления синдрома Рейно значительно уменьшились или исчезли.
Среди сосудорасширяющих препаратов эффективными средствами терапии синдрома Рейно являются антагонисты кальция. Нифедипин (коринфар, кордафен и ) назначают по 30-60 мг/сут. Для лечения синдрома Рейно можно использовать и другие блокаторы входа кальция: верапамил, дилтиазем, никардипин.
При прогрессирующем синдроме Рейно рекомендуется применение вазапростана (простагландин Еl, альпростадил). Вазапростан вводится внутривенно капельно в дозе 20-40 мкг альпростадила в 250 мл физиологического раствора в течение 2-3 часов через день или ежедневно, на курс 10-20 вливаний. Первоначальное действие вазапростана может проявиться уже после 2-3 вливания, но более стойкий эффект отмечается после окончания курса лечения и выражается в снижении частоты, продолжительности и интенсивности атак синдрома Рейно, уменьшении зябкости, онемения и болей. Положительное действие вазапростана обычно сохраняется в течение 4-6 месяцев, рекомендуется проводить повторные курсы лечения (2 раза в год).
Особое место в лечении сосудистых поражений занимают ингибиторы ангиотензинпревращающего фермента (АПФ), в частности каптоприл. Каптоприл назначается в дозе 25 мг 3 раза в день, рекомендуется длительное (6-12 месяцев) применение с индивидуальным подбором поддерживающих доз.
В лечении синдрома Рейно также используется кетансерин — селективный блокатор HS2-серотониновых рецепторов; назначается по 20-60 мг/сут, обычно хорошо переносится, может быть рекомендован пожилым больным.
Большое значение в лечении синдрома Рейно имеют препараты, улучшающие свойства крови, снижающие вязкость: дипиридамол по 75 мг и более в сутки; пентоксифиллин (трентал, агапурин) в дозе 800-1200 мг/сут внутрь и внутривенно; низкомолекулярные декстраны (реополиглюкин и ) — внутривенно капельно по 200-400 мл, на курс 10 вливаний.
При лечении синдрома Рейно следует учитывать необходимость длительной многолетней терапии и нередко комплексное применение препаратов разных групп.
Лекарственную терапию синдрома Рейно рекомендуется сочетать с применением других методов лечения (гипербарическая оксигенация, рефлексотерапия, психотерапия, физиотерапия).
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник