Синдром гипофизарной карликовости с нейросенсорной глухотой

Гипоталамо-гипофизарная карликовость с глухотой. Мукополисахаридозы: синдром ГурлерО 2 сестрах с гипоталамо-гипофизарной карликовостью и нейросенсорной глухотой сообщили Winkelmann с сотр. Отставание в росте впервые было замечено при поступлении в школу. У взрослых сестер рост был равен 139 и 146 см. Эндокринная система. Ни одна из девочек не достигла половой зрелости, ни у той, пи у другой не появились волосы на лобке и не развились молочные железы. У обеих сестер отмечались первичная аменорея и инфантилизм наружных и внутренних гениталий. Вестибулярная система. Результаты исследований не опубликованы. Радиоиммунологическое исследование уровня гормона роста в плазме крови показало резкое его снижение (3,0 нг/мл против 30 нг/мл в норме). При стимуляции инсулином уровень гормона роста повышался, но не выше чем до 5,0 нг/мл. Содержание фолликулин-стимулирующего (FSH) и лютеинизируюшего (LH) гормонов соответствовало препубертатному уровню. Усвоение радиоактивного йода, а также выделение кортизола и кортикостерона были нормальными. Уровень 17-гидрокси- и 11-гидрокси-кортикостероидов в моче был нормальным, даже после стимуляции АКТГ.
Мукополисахаридозы: синдромы Гурлер и ШейеМукополисахаридозы — наследственные болезни мукополисахаридного обмена. Дефект активности различных генетически контролируемых путей лизосомалыюй деградации ведет к внутриклеточному накоплению недеградированных кислых мукополисахаридов и к относительно схожим клиническим и скелетным изменениям. Фенотип наиболее резко выражен при синдромах Гурлер и Марото— Лами и менее резко при других мукополисахаридозах (McKusick, Spranger). В связи с малым объемом этого текста мы сможем только кратко в общих чертах представить клинические и лабораторные данные о тех мукополисахаридозах, которые сопровождаются глухотой. При синдроме Шейе (МПС-1-Ш), аллельной форме синдрома Гурлер, черты лица несколько грубоваты, наблюдаются нижнечелюстной прогнатизм и опущенные углы рта. Нос широкий. Начинающееся в молодости прогрессирующее помутнение роговицы приводит обычно к снижению остроты зрения в четвертом десятилетии жизни. Больные слегка отстают по росту от сверстников. Интеллект нормальный. Кисти и стопы широкие, пальцы па руках и ногах фиксированы в «когтеподобном» положении. Подвижность всех суставов ограничена. Часто наблюдается синдром «туннеля запястья». У большинства больных отмечается регургитация аорты. — Также рекомендуем «Синдромы Хантер, Санфилиппо, Моркио, Марото — Лами. Орган слуха при мукополисахаридозах» Оглавление темы «Наследственные болезни с глухотой»:
|
Источник
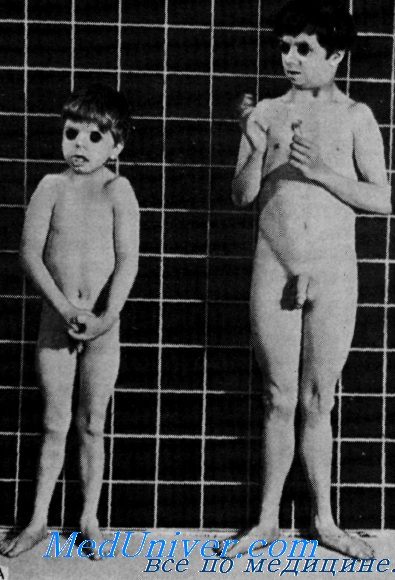
Пренатальное нарушение роста, с повышенным уровнем гормона роста и умственная отсталость с глухотойVan Gemund, Laurent de Angulo и van Gelderen сообщили о 2 мальчиках сибсах с пренаталыюй карликовостью, повышением в сыворотке крови иммунореактивного гормона роста и отсутствием реакции на пего организма, умственной отсталостью и врожденной глухотой. Клинические данные. Данные осмотра. У сибсов отмечалась внутриутробная задержка роста (при рождении в срок вес был менее чем 1900 г), но конечности и туловище были пропорциональными. Голова была несколько малых размеров. Признаки полового развития не выявлялись до 20-летнего возраста, даже под влиянием терапии. Орган слуха. У обоих сибсов была врожденная глухота и плохо развитая речь. Нарушения слуха подробно не описаны. Лабораторные данные. Рентгенологическое исследование показало резкую задержку созревания скелета. В сыворотке крови было обнаружено повышение уровня иммупореактивного гормона роста. Введенный экзогенно человеческий гормон роста вызывал нормальную реакцию в сыворотке крови уровней инсулина, глюкозы и свободных жиров, но недостаточное увеличение ретенции азота и выделения с мочой гидроксипролина.
Наследственность. Родители 2 больных мальчиков были здоровы и состояли в кровно-родственном браке. Заболевание, по-видимому, наследуется по аутосомно-рецессивному типу. Диагноз. Laron описал карликовость с высоким содержанием в сыворотке крови иммупореактивного человеческого гормона роста. На экзогенное введение гормона роста обследованные лица реагировали нормально. Najjar сообщил о сибсах, у которых ни при пероральном, ни при внутривенном введении глюкозы уровень человеческого гормона роста не снижался. Экзогенный гормон роста недостаточно повышал ретенцию азота и экскрецию с мочой гидроксипролина. Ни в одной семье не отмечалось умственной отсталости или глухоты. Лечение. Росту может способствовать терапия анаболическими стероидами. Прогноз. Хотя заболевание не угрожает жизни, прогноз неблагоприятный, так как лечение неэффективно. Выводы. Характеристика этого синдрома включает: — Также рекомендуем «Гипоталамо-гипофизарная карликовость с глухотой. Мукополисахаридозы: синдром Гурлер» Оглавление темы «Наследственные болезни с глухотой»:
|
Источник
Болезнь Тея — Сакса с глухотой. Молочная ацидемия, миопатия и низкий рост с глухотойМы полагали, что твердые данные о нарушениях слуха при разных мукополисахаридозах малочисленны. Рискуя прослыть придирчивыми, мы должны сказать, что еще меньше доступной информации имеется о нарушениях слуха при многих муколипидозах и сфинголипидозах. Обычно недостаток этих сведений является следствием того, что указанные заболевания часто сопровождаются глубоким слабоумием и ранней глухотой. Болезнь Тея — Сакса, согласно данным Kelemen, представившего обзор литературы и изучившего лично 2 случая, нередко сопровождается гиперакузией и средним отитом. Это выдержка из работы Boies. У больных GM1-ганглиозидозом, тип I, наблюдается нейросенсорная глухота (R. Desnick, личное сообщение). Goldberg с соавт. обнаружили двустороннюю нейросенсорную глухоту при болезни накопления, характеризующейся задержкой роста, грубыми чертами лица, умственной отсталостью, судорожными припадками, помутнением роговицы, вишнево-красным пятном на глазном дне, множественными дизостозами, дефицитом р-галактозидазы и аутосомно-рецессивным наследованием. Согласно нашим наблюдениям, у больных с муколипидозом-III часто выявляется легкая проводящая глухота.
Молочная ацидемия, миопатия и низкий рост с глухотойHackett, Bray, Ziter, Nyhan и Creer описали 2 сестер с этим, несомненно уникальным, синдромом. У обеих сестер отмечался низкий рост (ниже 3 S.D.). Нервная система. Интеллект был нормальным. У одной из сестер проявилась светостимулирующаяся эпилепсия. Орган слуха. У обеих сестер отмечалась умеренная нейросенсорная глухота, более подробно не охарактеризованная. Лабораторные данные. В моче и в крови у больных было обнаружено высокое содержание аланина. Нагрузочная проба с пищевым введением аланина выявила снижение почечного клиренса аланина. В крови отмечалось заметное повышение уровня пировиноградной и молочной кислот. В моче обнаружено низкое содержание креатина, но уровень креатин-фосфокиназы в сыворотке крови был нормальным. Патология. При люминесцентной микроскопии в скелетных и в сердечной мышцах были обнаружены очаги «гранулярного некроза». Ультраструктурное исследование этих областей выявило многочисленные большие митохондрии с дегенеративно измененными миофибриллами (D’Agostino et al.). Диагноз. Это заболевание имеет некоторое сходство с синдромом лрогрессирующей наружной офтальмоплегии с пигментной дегенерацией сетчатки, дефектом сердечной проводимости и смешанной глухотой и несколькими заболеваниями, обсуждаемыми в диагностическом плане с этим синдромом. Прогноз. Прогноз, несомненно, неблагоприятный. У одной из сестер молочный ацидоз привел к летальному исходу. Выводы. Синдром характеризуется: — Также рекомендуем «Гиперпролинемия и гиперпролинурия (иминоглицинурия) с глухотой» Оглавление темы «Наследственные болезни с глухотой»:
|
Источник
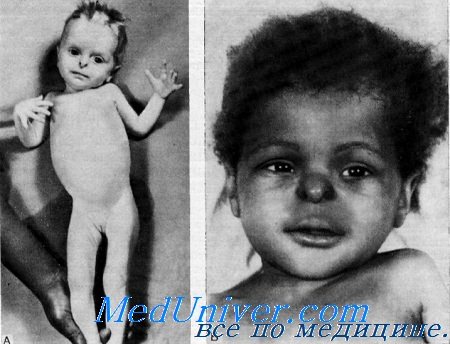
Аплазия крыльев носа, гипотиреоидизм, задержка роста и отсутствие зубов с глухотойJohanson и Blizzard и Park, Johanson, Jones, Blizzard выявили у 3 девочек из неродственных семей синдром, состоящий из аплазии крыльев носа, задержки роста, нарушения всасывания, гипотиреоидизма, отсутствия постоянных зубов и нейросенсорной глухоты. Morris и Fisher, а также Townes привели случаи болезни у женщин, по описали их недостаточно подробно. Клинические данные. Данные осмотра. Внешность больных производит странное впечатление из-за отсутствия крыльев носа. Переносье при этом может быть запавшим. У некоторых больных наблюдались срединные дефекты кожи волосистой части головы. При рождении вес обычно был небольшим. В раннем детстве больные также плохо прибавляли в весе. Па кистях и ногах отмечались отеки от умеренно выраженных до генерализованной анасарки. Если эти отеки не лечить, то может развиться застойная сердечная недостаточность. Дети обычно резко отставали в развитии (Johanson a. Blizzard). Однако у некоторых больных интеллект был нормальным (Townes). Мочеполовая система. Как правило, у больных имеется единственное мочеполовое отверстие. Обнаружены двойное влагалище и двойная матка с нормальными трубами и яичниками. Половое созревание задержано (Park et al.). Иногда наблюдается увеличенный клитор с хорошо сформированной препуциалыюй складкой. Костно-мышечная система. Обычно отмечаются мышечная гипотония и чрезмерная подвижность суставов. Костный возраст резко задержан. У большей части больных наблюдается микроцефалия. У одной больной обнаружено некоторое утолщение костей свода черепа. Окостенение эпифизов головки бедра, средних надмыщелков плеча, дистального эпифиза бедра и проксимального эпифиза большой берцовой кости протекает неправильно (Johanson a. Blizzard). Эти изменения, однако, не наблюдались в других опубликованных случаях. Постоянные зубы никогда не развиваются, кроме первых нижних коренных зубов, которые могут наблюдаться в виде редкого исключения. Эндокринная система. В большинстве случаев, если не во всех, наблюдался атпреоидный кретинизм.
Вестибулярная система. Результаты исследований не описаны. Он был низкого роста, с врожденной нейросенсорной глухотой, молочными зубами, гипоплазией крыльев носа, умственной отсталостью и легкой гипоспадией. Кроме того, у него отмечались микрофтальм и аниридия. Его кариотип был нормальным. Диагноз. Недостаточность трипсиногена сочетается с резким нарушением роста, жидким поносом и изъязвляющимся отеком конечностей, но другие аномалии, характерные для обсуждаемого здесь синдрома, не наблюдаются (Townes). Park с соавт. обследовали женщину с псевдогермафродитизмом, а также различными аномалиями половой системы и заднепроходного отверстия, по не имевшую атиреоидного кретинизма, задержки роста, глухоты, нарушении функции поджелудочной железы и олигодонтии, т. е. всех тех симптомов, которые наблюдаются при данном синдроме. Лечение. Дефект носа может быть исправлен при помощи ринопластинки. Дети должны строго соблюдать режим и принимать в дополнение к диете гидролизат белка, панкреатин и витамины. Атиреоидный кретинизм можно лечить при помощи L-тироксина или препаратом высушенной щитовидной железы. Наряду с улучшением роста отмечается неэффективность гормонотерапии для мышечной гипотонии и умственной отсталости. Выводы. Данный синдром характеризуется: — Также рекомендуем «Пренатальное нарушение роста, с повышенным уровнем гормона роста и умственная отсталость с глухотой» Оглавление темы «Наследственные болезни с глухотой»:
|
Источник