Синдром франческетти по мкб 10

Синдром Тричера Коллинза – Франческетти
Синдром Тричера Коллинза – Франческетти (мандибуло-фациальный дизостоз, челюстно-лицевой дизостоз, синдром Томпсона, TCOF, МКБ 10 – Q75.4) — редкое аутосомно-доминантное заболевание, для которого характерно нарушение черепно-лицевого развития. Проявляется рядом врожденных челюстно-лицевых дефектов, в их числе арковидное нёбо или расщелина нёба, большая ротовая щель и отсутствие ресниц на нижнем веке, дефект слухового прохода, приводящий к кондуктивной тугоухости. Молекулярно-генетической причиной заболевания являются мутации гена TCOF1, реже — генов POLR1C или POLR1D. Лечение включает хирургические методы, слухопротезирование и регулярные занятия с сурдопедагогом.
Впервые болезнь описал в 1846 г. британский врач А. Томпсон (A. Thompson). В 1900 г. британский хирург Эдвард Тричер Коллинз (Edward Treacher Collins) представил на заседании офтальмологического общества в Лондоне двух пациентов с характерными для болезни симптомами [1]. Термин «мандибуло-фациальный дизостоз» предложил в 1944 г. швейцарский офтальмолог Адольф Франческетти (Adolphe Franceschetti).
В 1996 г. было установлено, что молекулярно-генетической причиной заболевания в большинстве случаев является мутация в гене TCOF1 [6].
Распространенность и тип наследования
Для пациентов с синдромом Тричера Коллинза – Франческетти характерен ряд врожденных челюстно-лицевых дефектов. Это антимонголоидный разрез глаз (раскосые глаза, направленные вниз), гипоплазия нижней челюсти, дефект нижних век, аномалии ушных раковин, дефект слухового прохода, приводящий к кондуктивной тугоухости, двусторонняя гипоплазия скуловых костей и орбит, арковидное нёбо или расщелина нёба, большая ротовая щель и отсутствие ресниц на нижнем веке [4]. Лицевые кости у большинства пациентов слаборазвитые. Это приводит к формированию «затонувшего» типа лица: крупный нос и очень маленькие челюсти и подбородок. У некоторых больных присутствует «волчья пасть». В ряде случаев отмечаются рост волос на щеках, дефект верхнего века и радужной оболочки глаз.
Диагноз может быть установлен как во время беременности, так и после рождения. Диагностика для пациентов с характерным внешним видом включает исследование слуха, компьютерную томографию височных костей, анализ мутаций генов TCOF1, POLR1C и POLR1D [5].
Лечение синдрома Тричера Коллинза – Франческетти включает в себя хирургические операции, направленные на устранение дефектов лица (с целью улучшения внешнего вида и качества жизни пациента), стоматологическое лечение (в том числе ортодонтическое, направленное на исправление прикуса). Пациентам имплантируют слуховые аппараты костного проведения звука. Детям с мандибуло-фациальным дизостозом необходимы регулярные занятия с сурдопедагогом и логопедом.
Прогноз для пациентов, как правило, благоприятный. Важно как можно раньше провести слухопротезирование, чтобы избежать отставания ребенка в речевом и общем развитии. Существенную роль играют также регулярные занятия с сурдопедагогом и психологическая поддержка родителей ребенка [3].
Молекулярно-генетической причиной заболевания в 78–93 % случаев является мутация в гене TCOF1, кодирующем ядерный транспортный фосфопротеин. Этот белок принимает участие в транскрипции ДНК. Ген TCOF1 представлен во многих тканях организма во время эмбрионального и постэмбрионального развития. Нонсенс-мутация в гене TCOF1, приводящая к возникновению преждевременного стоп-кодона, ведет к нарушению синтеза белка. В результате вырабатывается недостаточное для нормального функционирования организма количество ядерного транспортного фосфопротеина [2], что способствует нарушению черепно-лицевого развития.
В 8 % случаев мандибуло-фациальный дизостоз связан с мутациями в генах POLR1C или POLR1D, кодирующих субъединицы I и III РНК-полимеразы. При мутации генов POLR1C или POLR1D происходит нарушение процесса транскрипции ДНК из-за недостаточного количества фермента РНК-полимеразы, что также способствует нарушению черепно-лицевого развития.
- Treacher Collin E, Cases with symmetrical congenital notches in the outer part of each lid and defective development of the malar bones, 1900, Trans Ophthalmol Soc UK, 20, p. 190—192
- Chiara Conte, Maria Rosaria D’Apice, Fabrizio Rinaldi, Stefano Gambardella, Federica Sangiuolo and Giuseppe Novelli (2011 Sep 27). Novel mutations of TCOF1 gene in European patients with Treacher Collins syndrome. BMC Med Genet. 12
- Козлова С. И., Демикова Н. С. Наследственные синдромы и медико-генетическое консультирование. – М.: КМК, 2007 – 448 с.
- Кеннет Л. Джонс Наследственные синдромы по Девиду Смиту. Атлас-справочник. Москва, Практика, 2011
- Кадышев В. В., Бессонова Л. А., Зинченко Р. А. Распространенность синдрома Тричер Коллинз-Франческетти в Кировской области. Медицинская генетика, 2009 г. Том 8. №12 (90) , 2009
- Treacher Collins Syndrome Collaborative Group. Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome. Nature Genet. 12: 130-136, 1996
- База данных Orphanet
Источник
Медицинский эксперт статьи

х
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
При внутриутробных нарушениях процессов развития костей возникают серьезные черепно-лицевые деформации, и одной из разновидностей такой патологии является синдром Тричер Коллинза (TCS) или мандибулофасциальный, то есть челюстно-лицевой дизостоз.
Код заболевания по МКБ 10: класс XVII (врожденные аномалии, деформации и хромосомные нарушения), Q75.4 — mandibulofacial dysostosis.
Код по МКБ-10
Q75.4 Челюстно-лицевой дистоз
Эпидемиология
Распространенность синдрома Тричер Коллинза находится в диапазоне – один случай на 25- 50 тыс. новорожденных обоего пола (хотя британские медики отмечают, что патология выявляется у одного младенца из 10-15 тыс.).
[1], [2], [3], [4], [5]
Причины синдрома Тричер Коллинза
Данный синдром получил имя выдающегося британского офтальмолога Эдварда Тричер Коллинза, описавшего основные черты патологии более ста лет назад. Однако европейские врачи чаще называют этот вид аномалии костей лица и челюстей болезнью или синдромом Франческетти – на основании обширных исследований швейцарского офтальмолога Адольфа Франческетти, который ввел термин «мандибулофасциальный дизостоз» в середине прошлого века. В медицинских кругах также используется название – синдром Франческетти-Коллинза.
Причины синдрома Тричер Коллинза – мутации гена TCOF1 (в локусе хромосомы 5q31.3-33.3), который кодирует ядрышковый фосфопротеин, отвечающий за формирование черепно-лицевой части эмбриона человека. В результате преждевременного уменьшения количества этого белка нарушаются биогенез и функции рРНК. По мнению генетиков исследовательской программы Human Genome, эти процессы приводят к сокращению пролиферации эмбриональных клеток нервного гребня – валика вдоль нервного желоба, который в ходе развития зародыша замыкается в нервную трубку.
Формирование тканей лицевой части черепа происходит благодаря трансформации и дифференциации клеток верхней (головной) части нервного гребня, которые мигрируют вдоль нервной трубки в область первой и второй жаберных дуг зародыша. И дефицит этих клеток вызывает черепно-лицевые деформации. Критический период возникновения аномалий – с 18 по 28 день после оплодотворения. По завершении миграции клеток нервного гребня (на четвертой неделе гестации) образуются практически все рыхлые мезенхимальные ткани в области лица, которые позже (с 5 по 8 недели) дифференцируются в скелетные и соединительные ткани всех частей лица, шеи, гортани, уха (в том числе внутреннего) и будущих зубов.
[6], [7], [8], [9]
Патогенез
Патогенез синдрома Тричер Коллинза часто имеет семейный характер, и аномалия наследуется по аутосомно-доминантному принципу, хотя бывают случаи аутосомно-рецессивной передачи дефекта (при мутациях других генов, в частности, POLR1C и POLR1D). Самым непредсказуемым в челюстно-лицевом дизостозе является то, что мутация наследуется детьми только в 40-48% случаев. То есть у 52-60% пациентов причины синдрома Тричер Коллинза не связаны с наличием аномалии в роду, и, как полагают, патология возникает в результате спорадических генных мутаций de novo. Вероятнее всего, новые мутации представляют собой последствия тератогенного воздействия на плод во время беременности.
В числе тератогенных причин данного синдрома специалисты называют большие дозы этанола (этилового спирта), радиацию, сигаретный дым, цитомегавирус и токсоплазму, а также гербициды на основе глифосата (Раундал, Глифор, Торнадо и др.). А в список ятрогенных факторов попали препараты от угрей и себореи с 13-цис-ретиноевой кислотой (Изотретиноин, Аккутан); противосудорожное лекарство Фенитоин (Дилантин, Эпанутин); психотропные средства Диазепам, Валиум, Реланиум, Седуксен.
[10], [11], [12], [13], [14], [15], [16], [17], [18]
Симптомы синдрома Тричер Коллинза
По большей части, клинические признаки мандибулофасциального дизостоза и степень их выраженности зависят от особенностей проявления генных мутаций. И первые признаки данной аномалии в большинстве случаев видны у ребенка сразу же после его появления на свет: лицо при синдроме Тричер Коллинза имеет характерный вид. Причем морфологические аномалии обычно двусторонние и симметричные.
Наиболее очевидные симптомы синдрома Тричер Коллинза:
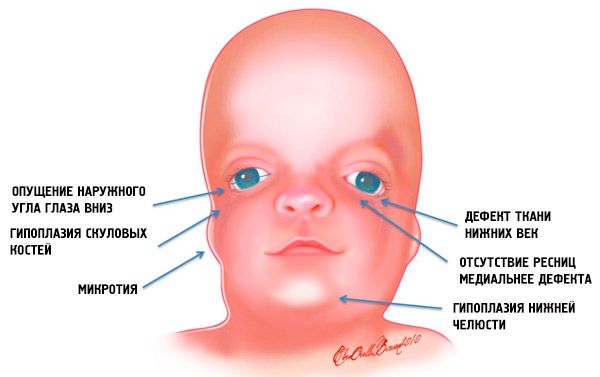
- недоразвитость (гипоплазия) лицевых костей черепа: скуловых, скуловых отростков лобной кости, боковых крыловидных пластинок, придаточных пазух носа, нижней челюсти и выступов костных эпифизов (мыщелков);
- недоразвитие костей нижней челюсти (микрогнатия) и более тупой чем обычно нижнечелюстной угол;
- нос имеет нормальный размер, однако, кажется большим из-за гипоплазии надбровных дуг и недоразвитости или отсутствия скуловых дуг в области висков;
- глазные щели нисходящие, то есть разрез глаз аномальный, с опущенными вниз наружными уголками;
- дефекты нижних век (колобома) и частичное отсутствие ресниц на них;
- ушные раковины неправильной формы с широким диапазоном отклонений, вплоть до их расположения в углу нижней челюсти, отсутствия мочек, слепых свищей между козелком уха и углом рта и др.;
- сужение или заращение (атрезия) наружного слухового каналов и аномалии косточек среднего уха;
- отсутствие или гипоплазия околоушных слюнных желез;
- фарингеальная гипоплазия (сужение глотки и дыхательных путей);
- несращение твердого нёба (волчья пасть), а также отсутствие, укорочение или неподвижность мягкого неба.
Такие анатомические аномалии во всех случая имеют осложнения. Это функциональные нарушения слуха в виде проводящей (кондуктивной) тугоухости или полной глухоты; нарушения зрения из-за неправильно формирования глазных яблок; дефекты нёба вызывают трудности с кормлением и глотанием. Имеются связанные с дефектами челюстей нарушения окклюзии зубов (неправильный прикус), что, в свою очередь, вызывает проблемы с жеванием и артикуляцией. Патологии мягкого неба объясняют гнусавость голоса.
Осложнения и последствия
Последствия челюстно-лицевых аномалий при синдроме Тричер Коллинза проявляются в том, что при рождении ребенка его интеллектуальные способности нормальные, но из-за дефектов слуха и других нарушений отмечается вторичная задержка умственного развития.
Кроме того, дети с такими дефектами остро чувствуют свою ущербность и страдают, что негативно сказывается на их нервной системе и психике.
[19], [20], [21]
Диагностика синдрома Тричер Коллинза
Постнатальная диагностика синдрома Тричер Коллинза, по существу, проводится на основании клинических признаков. Челюстно-лицевой дизостоз легко определяется при полный экспрессивности синдрома, но когда присутствуют минимально выраженные симптомы патологии, с постановкой правильного диагноза могут возникнуть проблемы.
При этом особого внимания требует оценка всех связанных с аномалиями функций, особенно тех, что затрагивают дыхание (в связи с угрозой апноэ во сне). Также проводится оценка и мониторинг эффективности кормления и насыщения гемоглобина кислородом.
В дальнейшем — на 5-6 день после рождения — предстоит выяснить степень повреждений слуха с помощью аудиологического тестирования, которое должно проводиться еще в родильном доме.
Назначается обследование, в ходе которого инструментальная диагностика проводится рентгеноскопией черепно-лицевой дисморфологии; пантомографией (панорамным рентгеном костных структур лицевого черепа); полной черепной компьютерной томографией в различных проекциях; КТ или МРТ головного мозга для определения состояния внутреннего слухового прохода.
Самое раннее – пренатальное – диагностирование челюстно-лицевых аномалий при наличии синдрома Тричер Коллинза в семейном анамнезе возможно путем биопсии ворсин хориона на 10-11 неделе беременности (процедура угрожает выкидышем и занесением инфекции в матку).
Также берутся анализы крови членов семьи; на 16-17 неделе беременности берется анализ околоплодных вод (трансабдоминальный амниоцентез); на 18-20 неделях беременности проводится фетоскопия и берется кровь из плодовых сосудов плаценты.
Но чаще всего в дородовой диагностике данного синдрома у плода используется УЗИ (на 20-24 неделях беременности).
[22], [23]
Какие анализы необходимы?
Дифференциальная диагностика
Этими же методами специалисты пользуются, когда нужна дифференциальная диагностика, чтобы распознать неярко выраженный синдром Тричер Коллинза и отличить его от других врожденных аномалий черепно-лицевых костей, в частности: синдромов Апера, Крузона, Нагера, Петерс-Хевельса, Хеллермана-Штефа, а также с гемифациальной микросомии (синдрома Гольденхара), гипертелоризма, преждевременного заращения швов черепа (краниостеноза) или нарушения сращения лицевых костей (краниосиностоза).
[24], [25], [26], [27]
Лечение синдрома Тричер Коллинза
Как и во всех случаях, генетически обусловленных врожденных дефектов, лечение синдрома Тричер Коллинза в тяжелых формах носит исключительно паллиативный характер, поскольку терапевтических методов при таких патология просто не существует. Спектр и степень деформаций при данном синдроме обширны и, следовательно, характер и интенсивность врачебного вмешательства также имеет множество вариантов.
Для коррекции и улучшения слуха используются слуховые аппараты, для улучшения речи – занятия с логопедом.
Хирургические вмешательства требуются в раннем возрасте в тяжелых случаях сужения дыхательных путей (проводят трахеостомию) и гортани (выполняется гастростоммия для кормления) Также может потребоваться оперативная коррекция нёба.
Операции по удлинению нижней челюсти выполняются в возрасте 2-3 лет или позже. Реконструкция мягких тканей включает в себя коррекцию колобомы нижнего века и пластику ушных раковин.
Профилактика
Профилактика синдрома Тричер Коллинза состоит в посещении будущими родителями генетической консультации, а при семейном анамнезе синдрома возникает вопрос о возможности самой беременности – во избежание рождения ребенка с черепно-лицевыми аномалиями.
[28], [29], [30], [31]
Прогноз
Каким может быть прогноз при данной патологии? Это зависит от степени деформаций и интенсивности симптоматики. Синдром Тричер Коллинза – это пожизненный диагноз.
[32], [33], [34], [35]
Источник
Синдром Тричера Коллинза (англ. Treacher Collins syndrome, TCS, челюстно-лицевой дизостоз) — аутосомно-доминантное заболевание, характеризующееся черепно-лицевой деформацией. Описан английским офтальмологом Эдвардом Тричером Коллинзом в 1900 году[1].
Синдром Тричера Коллинза встречается у 1 из 50 000 младенцев[2]. Типичные клинические признаки: грубый дефект лицевой части черепа, косоглазие, колобомы век; размер рта, подбородка и ушей существенно меньше нормы. В некоторых случаях — ослабление слуха.
Этиология[править | править код]
Причиной заболевания является, чаще всего, нонсенс-мутация (возникновение стоп-кодона) в гене TCOF1, приводящая к гаплонедостаточности.
Ген TCOF1 расположен в локусе 5q32-33, на длинном (q) плече 5-й хромосомы, начинается от 149 737 202-й пары оснований и заканчивается на 149 779 871-й паре оснований[3][4].
Продукт гена TCOF1 — ядерный транспортный белок, который экспрессируется во многих тканях во время эмбрионального и постэмбрионального развития и принимает участие в транскрипции ДНК. При синдроме Тричера Коллинза развивается состояние, при котором половинного количества генного продукта недостаточно для нормального функционирования организма[5].
Синдром наследуется по аутосомно-доминантному принципу и характеризуется высокой пенетрантностью. Экспрессивность может быть от умеренной до выраженной, потому тяжесть дефекта отлична у разных пациентов — от почти незаметных признаков до крайне тяжёлых форм. У большинства пациентов слаборазвитые лицевые кости, что приводит к «затонувшему» лицу, крупный нос и очень маленькие челюсти и подбородок (микрогнатия). У некоторых больных присутствует волчья пасть. В тяжелых случаях микрогнатия может вытеснять язык пострадавших новорожденных достаточно, чтобы вызвать преграду ротоглотки и потенциально опасных для жизни заболеваний дыхательных путей. Врождённый порок сердца является необычной особенностью[6].
Лечение[править | править код]
В лечении пациентов, пострадавших от ТКС, применяется междисциплинарный подход, то есть требуются вмешательства различных специалистов. Основные проблемы у пациентов с ТКС — нарушения глотания и проходимости дыхательных путей. Некоторые пациенты нуждаются в трахеостомии. Гастростома может быть необходима для обеспечения адекватного потребления пищи и для защиты дыхательных путей[7]. Вопрос о хирургическом восстановлении структуры лица решается индивидуально и осуществляется по достижении определённого возраста[8].
Потеря слуха при синдроме Тричера Коллинза вызвана деформацией структур в наружном и среднем ухе. Потеря слуха, как правило, двусторонняя. Даже в тех случаях, когда ушные раковины и наружные слуховые проходы не затронуты синдромом, цепи слуховых косточек часто повреждены[9].
Попытки хирургическим путём восстановить наружный слуховой проход для улучшения слуха у детей с ТКС не дали положительных результатов[10]. Предпочтительной оказалась слуховая реабилитация со слуховыми аппаратами костной проводимости[en] (BAHA) или обычными слуховыми аппаратами[11].
В культуре[править | править код]
- «Чудо» — художественный фильм-драма 2017-го года с главным героем с синдромом ТК
Примечания[править | править код]
- ↑ Treacher Collin E, «Cases with symmetrical congenital notches in the outer part of each lid and defective development of the malar bones», 1900, Trans Ophthalmol Soc UK, 20, p. 190—192
- ↑ Chiara Conte, Maria Rosaria D’Apice, Fabrizio Rinaldi, Stefano Gambardella, Federica Sangiuolo and Giuseppe Novelli. Novel mutations of TCOF1 gene in European patients with Treacher Collins syndrome (англ.) // BMC Med Genet. : journal. — 2011. — 27 September (vol. 12). — doi:10.1186/1471-2350-12-125. — PMID 21951868.
- ↑ Dixon M.J., Dixon J., Raskova D., et al. Genetic and physical mapping of the Treacher Collins syndrome locus: refinement of the localization to chromosome 5q32-33.2 (англ.) // Human Molecular Genetics (англ.)русск. : journal. — Oxford University Press, 1993. — Vol. 1, no. 4. — P. 249—253. — doi:10.1093/hmg/1.4.249. — PMID 1303194.
- ↑ Dixon M.J., Dixon J., Houseal T., et al. Narrowing the position of the Treacher Collins syndrome locus to a small interval between three new microsatellite markers at 5q32-33.1 (англ.) // American Journal of Human Genetics (англ.)русск. : journal. — 1993. — Vol. 52, no. 5. — P. 907—914. — PMID 8488840.
- ↑ Синдром Тричер Коллинза-Франческетти (мандибулофациальный дизостоз, TCOF) //Центр молекулярной генетики Медико-генетического научного центра РАМН
- ↑ Электронный научный журнал «Медицина и образование в Сибири»
- ↑ Goel L., Bennur S.K., Jambhale S. Treacher Collins syndrome-a challenge for anaesthesiologists (англ.) // Indian Journal of Anaesthesia (англ.)русск. : journal. — 2009. — August (vol. 53, no. 4). — P. 496—500. — PMID 20640217.
- ↑ Evans, Adele Karen; Rahbar, Reza; Rogers, Gary F.; Mulliken, John B.; Volk, Mark S. Robin sequence: A retrospective review of 115 patients (англ.) // International Journal of Pediatric Otorhinolaryngology : journal. — 2006. — 31 May (vol. 70, no. 6). — P. 973—980. — doi:10.1016/j.ijporl.2005.10.016. — PMID 16443284.
- ↑ Argenta, Louis C.; Iacobucci, John J. Treacher Collins Syndrome: Present concepts of the disorder and their surgical correction (англ.) // World Journal of Surgery (англ.)русск. : journal. — 1989. — 30 June (vol. 13, no. 4). — P. 401—409. — doi:10.1007/BF01660753. — PMID 2773500.
- ↑ Marres, HA; Cremers, CW; Marres, E.H. Treacher-Collins syndrome. Management of major and minor anomalies of the ear (англ.) // Revue de laryngologie — otologie — rhinologie : journal. — 1995. — Vol. 116, no. 2. — P. 105—108. — PMID 7569369.
- ↑ Marres, H.A. Hearing loss in the Treacher-Collins syndrome (неопр.) // Advances in oto-rhino-laryngology. — 2002. — Т. 61. — С. 209—215. — doi:10.1159/000066811. — PMID 12408086.
Ссылки[править | править код]
- «Новое лицо Джулианы» — документальный фильм о девочке, родившейся с синдромом Тричера Коллинза тяжёлой формы
Источник