Синдром фрагильной ломкой х хромосомы

Генетические особенности синдрома ломкой Х-хромосомы
Среди группы наследственных болезней есть два заболевания, относящихся к самым частым причинам интеллектуальной недостаточности. Самая известная и наиболее распространённая патология – синдром Дауна, связанный с наличием лишней 21-ой хромосомы в геноме человека. В этой статье мы расскажем о втором по распространенности наследственном заболевании, которое приводит к умственной отсталости, а также может сопровождаться другими клиническими проявлениями.
Синдром ломкой X-хромосомы или синдром Мартина-Белл является результатом нарушения в гене FMR1 (fragile X mental retardation-1), который расположен на Х-хромосоме и играет важную роль в появлении и развитии нервных связей, обучении и запоминании. Частота этого синдрома среди мальчиков составляет 1:4000.
Так называемая «ломкость» X-хромосомы проявляется в том, что хромосома выглядит нетипично при специальном окрашивании, как будто один кусок отделился, хотя физически она остается цельной. Генетическая основа этого явления заключается в увеличении числа тринуклеотидных повторов CGG в гене FMR1, расположенном на X-хромосоме.
У здоровых людей число повторов в этом гене колеблется от 5 до 54. Если повторов больше 200, то наработка белка с гена FMR1 нарушается, что приводит к развитию синдрома Мартина-Белл и клиническому проявлению заболевания. Премутационное состояние — это количество повторов CGG от 55 до 200. В таком состоянии заболевание у людей в типичной форме не проявляется, но чем больше повторов в этом гене у носителя, тем больше вероятность того, что у ее или его детей количество повторов будет больше 200 и заболевание разовьется. В случае носительства премутации при формировании половых клеток количество повторов может увеличиваться, поэтому если у родителя количество повторов от 55 до 200, то высока вероятность рождения ребенка с мутантным геном FMR1 и синдромом Мартина-Белл. При этом носительство премутационного состояния будущим папой и мамой неравнозначно по вероятности возникновения мутантного аллеля у их детей: если носитель – мама, то вероятность значительного увеличения числа повторов гораздо выше. Количество повторов от 45 до 54 является промежуточной формой, которая не имеет никакого влияния на здоровье человека, но может приводить к проблемам у будущих поколений, как и в случае премутационного состояния гена.
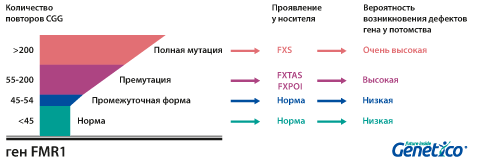
Важно учитывать, что наследование и развитие заболевания зависит от пола, так как ген FMR1 находится на Х-хромосоме. У мужчин только одна Х-хромосома, которую они получают от матери. Поэтому, в случае, если эта одна хромосома оказалась «ломкой», у них проявляется заболевание. У женщин две Х-хромосомы, однако активно работает только одна из них. Поэтому наличие одной Х-хромосомы с мутантным геном FMR1 может не проявляться клинически, в случае инактивации именно «ломкой» хромосомы, или приводить к развитию заболевания в 30-50% случаев. Мужчина с ломкой Х-хромосомой может передать её всем дочерям, но ни одному из сыновей. Женщина с мутантной хромосомой имеет шансы передать её как сыновьям, так и дочерям с равной вероятностью.
Премутационное состояние гена влияет как на судьбу потомков носителя такого гена, так и непосредственно на его здоровье:
Развитие первичной недостаточности яичников (FXPOI) (снижение овариального резерва и наступление менопаузы до 40 лет). Мутация FMR1 является причиной преждевременного истощения яичников у 5% женщин с этим диагнозом. Среди носительниц премутации примерно у четверти развивается это состояние. Оно влияет не только на общие репродуктивные возможности, но и на подбор протокола стимуляции при ВРТ, так как часто оказывается причиной бедного ответа яичников на стимуляцию. Интересно, что по данным, полученным в центре Genetico, хотя бедный ответ яичников на стимуляцию влияет на число получаемых в цикле эмбрионов, он не приводит к увеличению доли анеуплоидных эмбрионов.
Тремор/атаксия, ассоциированные с ломкой Х-хромосомой (FXTAS). Это состояние чаще развивается у мужчин: при носительстве премутации мужчиной проявляется в 33% случаев, а при носительстве премутации женщиной – лишь в 5-10%. Синдром FXTAS начинает проявляться в пожилом возрасте. Наблюдается тремор, шаткая походка, может страдать речь.
Метод диагностики, используемый в лаборатории Genetico, основан на использовании полимеразной цепной реакции с особым набором праймеров, позволяющих не только детектировать нормальное, премутационное и мутационное состояния, но и точно определить количество повторов в случаях, когда их меньше 200. Такая диагностика позволяет выявить синдром ломкой X-хромосомы на молекулярном уровне, а также оценить вероятность рождения ребенка с этим синдромом и возможность развития у пациента расстройств, связанных с увеличенным количеством повторов в гене FMR1. Такая диагностика также позволяет детектировать наличие AGG повторов среди повторов CGG. Полагают, что участки AGG, прерывающие длинную последовательность из CGG повторов, придают ДНК устойчивость и снижают риск увеличения количества повторов в следующем поколении.
Генетический тест, определяющий количество повторов в гене FMR1, рекомендуется пройти в первую очередь женщинам с синдромом преждевременного истощения яичников или с выявленной неслучайной инактивацией Х-хромосомы (косвенный признак), семьям, в которых есть сыновья с интеллектуальной недостаточностью. Также анализ состояния гена FMR1 необходим:
1) женщинам с репродуктивными проблемами или нарушениями фертильности, связанными с повышенным уровнем фолликулостимулирующего гормона (ФСГ)
2) пациентам с интеллектуальной недостаточностью и их родственникам
3) тем, у кого в семье были случаи синдрома ломкой Х-хромосомы или умственной отсталости без точного диагноза
4) женщинам, у родственников которых наблюдались нарушения, связанные с премутационным состоянием FMR1
5) пациентам с поздно проявившимся тремором и мозжечковой атаксией (нарушения согласованности работы мышц из-за поражения систем мозга, управляющих движением мышц).
В случае обнаружения бессимптомного носительства мутации в гене FMR1 у женщины может быть рекомендовано использование донорских ооцитов или проведение преимплантационной генетической диагностики (ПГД) с целью исключить возможность проявления синдрома у ребенка. Также важно правильно оценивать риск рождения больного ребенка в случае премутационного состояния гена FMR1 у будущих родителей. В таком случае по результатам теста рекомендуется консультация врача-генетика.
Автор: Очир Мигяев
Стажер лаборатории Genetico
Источник
Ломкие сайты хромосом, или фрагильные сайты (от англ. fragile — ломкий, хрупкий) — участки хромосом человека, склонные к образованию разрывов, которые выявляются при цитогенетическом анализе препаратов метафазных хромосом. Различают редкие, или наследуемые, и обычные, или конститутивные, ломкие сайты. Ломкие сайты имеются во всех хромосомах человека, в целом их насчитывается около сотни[1]. Молекулярная природа этого явления ещё не известна.
Номенклатура[править | править код]
Ломкие сайты обозначают в соответствии с тем, в каком хромосомном сегменте они находятся, например, ломкий сайт, ассоциированный с синдромом Мартина — Белл, имеет обозначение fra(X)(q27.3). Кроме того, существуют названия для ломких сайтов, утверждаемые комитетом по номенклатуре HUGO. Например, вышеупомянутый ломкий сайт fra(X)(q27.3) имеет название FRAXA, что означает «ломкий сайт на хромосоме X в локусе А», причём буква «А» означает, что это был первый описанный ломкий сайт для хромосомы Х[2].
Наследуемые сайты ломкости[править | править код]
Явление повышенной ломкости хромосом в определённых сайтах было обнаружено в 70-х годах XX века. При цитогенетическом анализе метафазных хромосом у некоторых индивидов было обнаружено, что в большинстве проанализированных клеток один и тот же участок хромосом имел разрыв или пробел в окрашивании. Частота встречаемости отдельных ломких сайтов в популяции не превышает обычно 5 %[2]. Для наследуемых ломких сайтов характерно менделевское наследование[1]. Выявлению большей части наследуемых ломких сайтов способствует культивирование клеток in vitro в среде, обеднённой фолиевой кислотой.
Большинство наследуемых фрагильных сайтов не связано с какой-либо клинически значимой патологией, кроме наследуемого сайта ломкости FRAXA, который наблюдается у больных синдромом хрупкой Х-хромосомы (Синдром Мартина — Белл). До развития молекулярно-генетических методов диагноз у пациентов с синдромом Мартина — Белл верифицировали по наличию ломкого сайта в локусе Xq27.3[3]. Наследуемый фрагильный сайт FRAXA находится в 5′-нетранслирумой области гена FMR1 и содержит повтор из триплетов ЦГГ. Аномальная длина этого повтора у больных вызывает гиперметилирование промотора гена FMR1 и, как следствие, нарушение экспрессии гена[4].
Конститутивные фрагильные сайты[править | править код]
Конститутивные фрагильные сайты — это разрывы и пробелы в окрашивании хромосом, которые появляются в определённых хромосомных сайтах в клетках у всех людей при умеренном репликативном стрессе, например, при применении в небольших концентрациях ингибиторов репликации ДНК[5]. Это гораздо более обширный класс фрагильных сайтов по сравнению с наследуемыми сайтами.
Конститутивные фрагильные сайты вызывают особенный интерес, потому что они являются «горячими точками» для хромосомных перестроек при различных раковых заболеваниях. Наиболее ярким примером является ломкий сайт FRA3B, располагающийся в хромосомном сегменте 3p14.2. Этот ломкий сайт находится в гене-супрессоре опухолевого роста FHIT[en], который часто утрачивается в опухолях различных локализаций, включая раки кишечника, головы-шеи, лёгких и раке молочной железы[2].
Природа явления ломкости хромосом до конца не изучена. Известно, что конститутивные фрагильные сайты обычно ассоциированы с позднореплицирующимся хроматином[6]. Они нередко находятся в пределах очень длинных генов (около 1 млн пар оснований и более), таких как FHIT и WWOX[en][7]. Многие обычные сайты ломкости являются тканеспецифичными. Недавние исследования связывают ломкость хромосом с дефицитом сайтов инициации репликации в этих районах. Предполагают, что недостаточность сайтов инициации в конце S-фазы клеточного цикла может приводить к локальной незавершенности процесса репликации при репликативном стрессе и формировании в некоторых случаях двунитевого разрыва ДНК[8].
Примечания[править | править код]
- ↑ 1 2 Durkin S. G., Glover T. W. Chromosome fragile sites. // Annu Rev Genet. — 2007. — Т. 41. — С. 169-192. — doi:10.1146/annurev.genet.41.042007.165900.
- ↑ 1 2 3 Генетика человека по Фогелю и Мотулски / М. Р. Спейчер, С. Е. Антонаракис, А. Г. Мотулски. — 4-е издание. — СПб: Н-Л. — С. 138-139. — 1056 с. — ISBN 978-5-94869-167-1.
- ↑ Oostra B. A. et al. Guidelines for the diagnosis of fragile X syndrome. National Fragile X Foundation (англ.) // Journal of medical genetics. — Vol. 30, no. 5. — P. 410-413.
- ↑ Naumann A. et al. A Distinct DNA-Methylation Boundary in the 5′-Upstream Sequence of the FMR1 Promoter Binds Nuclear Proteins and Is Lost in Fragile X Syndrome (англ.) // The American Journal of Human Genetics. — 2009. — Vol. 85, no. 5. — P. 606-616.
- ↑ Debatisse M. et al. Common fragile sites: mechanisms of instability revisited (англ.) // Trends in genetics : TIG. — 2012. — Vol. 28, no. 1. — P. 22-32.
- ↑ Wang L. et al. Allele-specific late replication and fragility of the most active common fragile site, FRA3B (англ.) // Human molecular genetics. — 1999. — Vol. 8, no. 3. — P. 431-437.
- ↑ Smith D. I. et al. Common fragile sites, extremely large genes, neural development and cancer //Cancer letters. – 2006. – V. 232. – №. 1. – P. 48-57.
- ↑ Letessier A. et al. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site //Nature. – 2011. – V. 470. – №. 7332. – P. 120-123.
Источник
Хромосомы фрагильной (ломкой) X синдром (синдром Мартина-Белла).
Это заболевание встречается в среднем у одного из 1200 мужчин и, возможно, 1 из 800 женщин. Это самая распространенная причина умственной отсталости, и по распространенности среди различных форм умственной отсталости она уступает только синдрому Дауна. Данное заболевание относят к моногенным, но закономерности наследования этой болезни необычны для Х-сцепленного признака.
В значительном числе случаев, от 20 до 40%, умственная отсталость у мальчиков передавалась от матери-носителя поврежденной Х-хромосомы.
В этих 20-40% мать-носитель получила свою поврежденную хромосому не от матери, как обычно, а от вполне внешне здорового отца. Вторая странность этой болезни — так называемый парадокс Шермана — заключается в различной степени пенетрантности мутации синдрома ломкости Х-хромосомы в зависимости от места носителя в родословной. Наконец, существует третья странность. Среди женщин-носителей мутантной хромосомы примерно треть оказывается в различной степени пораженной заболеванием, и, вдобавок, дети таких пораженных женщин с большей вероятностью оказываются больными, чем дети интеллектуально нормальных женщин-носителей. Эти больные женщины получают свою поврежденную Х-хромосому от матери, а не от отца. В целом, получается так, что дочери нормальных мужчин-передатчиков с большей вероятностью имеют больных детей, чем матери нормальных мужчин-передатчиков. Здоровые мужчины-передатчики передают свою поврежденную Х-хромосому дочерям, которые становятся носителями, но здоровы, а вот сыновья этих дочерей оказываются с высокой вероятностью больными (парадокс Шермана).
В определенных условиях культивирования клеток, полученных от пациентов с такими симптомами, на дистальном (удаленном от центромеры) конце длинного плеча Х-хромосомы (Xq28) отделялся фрагмент от ее основной части. Поэтому болезнь и была названа синдромом ломкости Х-хромосомы. Этот сайт на хромосоме называют FraXA от англ, fragile. Такого рода поведение различных хромосом достаточно хорошо известно, хотя причины его неясны. Все подобные сайты так и называются fragile, а тот, о котором идет речь, еще и ХА, потому что он расположен на Х-хромо- соме, но там есть еще и другие хрупкие сайты. Наблюдать этот цитогенетический эффект трудно.
В связи с этим был весьма понятен интерес к клонированию гена. Физическое выделение гена, ответственного за данную патологию, было осуществлено благодаря координированным усилиям множества групп. Им оказался ген FMR1. Было также установлено, что в развитии заболевания играет определяющую роль эффект так называемой динамической мутации. Относительно недавно был выявлен новый класс так называемых динамических мутаций, или мутаций экспансии, связанных с нестабильностью числа тринуклеотидных повторов в функционально значимых частях генов. Болезнь развивается лишь тогда, когда число повторов в этих сайтах превосходит определенный критический уровень. Наследование таких мутаций отличается от классического менделевского типа. Для них характерны: различная пенетрантность в сочетании с неполным доминированием; геномный импринтинг (различия фенотипических проявлений в зависимости от того, получена мутация от матери или от отца) и феномен антиципации — нарастание тяжести проявления заболевания в последующих поколениях. Этот тип мутаций пока найден только у человека и не зарегистрирован ни у одного вида млекопитающих или других хороню изученных организмов.
Классическим примером мутаций экспансии является синдром ломкой Х-хромосомы (FraXA), обусловленной присутствием удлиненных CGG-повторов в 5′-нетранслируемой регуляторной области FMR 1-гена (Xq27.3). В дальнейшем аналогичные динамические мутации были описаны и при 7 других наследственных заболеваниях, контролируемых генами, расположенными на разных хромосомах.
Причиной повреждающего действия одних «динамических» мутаций является блок генной экспрессии, то есть потеря функции (coss-of-function mutation), тогда как другие мутации того же типа, связанные с нейродегенеративными заболеваниями, ведут к появлению белковых продуктов с аномальными функциями (мутации типа gain-of-function). Для каждой болезни «экспансии» разработан свой вариант диагностики, основанный на полимеразной цепной реакции.
Еще по теме Хромосомы фрагильной (ломкой) X синдром:
- Хромосомы 18 трисомии синдром
- Хромосомы X моносомии синдром
- Хромосомы 1 3 трисомии синдром
- Хромосомы ХХУ синдром
- Хромосомы 21 трисомии синдром
- СИНДРОМ ДИСЕМІНОВАНОГО ВНУТРІШНЬОСУДИННОГО ЗГОРТАННЯ КРОВІ (ДВЗ-СИНДРОМ) В АКУШЕРСТВІ
- Синдром тестикулярної фемінізації (синдром нечутливості до дії андрогенів)
- Синдром надколенннково-бедренной боли (пателлофеморальный болевой синдром)
- Синдром Шерешевского—Тернера
- Синдром 47.ХУУ
- Наследственный нефрит. Синдром Альпорта
- Синдром Дауна
- Синдром трисомии X
- HELLP-синдром
- Нефротический синдром
- Синдром Кушинга
- ГІПЕРТЕРМІЧНИЙ СИНДРОМ
Источник
При
данной хромосомной аномалии рождение
живых детей не наблюдается, они погибают
на ранних стадиях эмбриогенеза.
Болезни, обусловленные хромосомными аберрациями Синдром «Крика кошки»
Впервые
синдром описан Леженом в 1964 г. (Франция).
Частота
встречаемости:
1:50000 новорожденных .
Причина:
делеция (отрыв) короткого плеча 5-й
хромосомы, с утратой от 1/3
до ½ короткого плеча. Болеют чаще
девочки.
Клиника:
синдром получил название от специфического
плача детей, напоминающего кошачье
мяуканье. Это обусловлено аномалиями
в строении гортани: узкая гортань,
уменьшенный надгортанник, мягкие хрящи,
необычные складки слизистой оболочки.
С возрастом этот симптом исчезает, но
остается склонность к простудным
заболеваниям верхних дыхательных путей.
Лицо у таких больных лунообразное, с
широко поставленными глазами. Отмечается
микроцефалия (уменьшенный размер мозга),
нередко — четырехпалостъ, пороки развития
сердечно-сосудистой системы,
желудочно-кишечного тракта, а также
аномалии почек (недоразвитие,
подковообразные почки, удвоение лоханок).
У всех имеется значительное снижение
интеллекта.
Продолжительность
жизни небольшая. Большинство умирает
в первые годы. Около 10% доживают до 10
лет.
Диагностика:
кариотипирование — укорочение короткого
плеча 5-й хромосомы, 46, 5р –; дерматоглифика
— поперечная складка на ладони.
Синдром «Филадельфийской» хромосомы
Впервые
описан в г. Филадельфии США в 1961 г.
Тооджем.
Причина:
делеция половины длинного плеча у 21-й
хромосомы, так считалось до 1970г. За
последние тридцать лет характер аберрации
уточнился – транслокация делетированного
участка длинного плеча 22 хромосомы на
длинное плечо 9-ой хромосомы, а небольшого
участка 9-ой на 22-ую – t
(9; 22) (q
34; q
11). При этом образуется структура,
обладающая онкогенными свойствами.
Клиника:
развивается хронический миелолейкоз,
что выражается в безудержном размножении
гранулоцитов (один из видов лейкоцитов),
в итоге в периферической крови появляется
много незрелых форм этих лейкоцитов.
Диагностика:
кариотипирование, обнаружение
соответствующей аберрации.
Синдром Мартина-Белла (иди синдром фрагильной х-хромосомы)
Частота
встречаемости
1:2000-4000 новорожденных.
Причина:
делеция (отрыв) небольшого концевого
(дистального) участка длинного плеча
Х-хромосомы, где располагается мутантный
ген ломкости. С помощью методов
молекулярно-генетического анализа в
нетранслируемой области гена FMR-I
(fragile
mental
retardation)
была обнаружена экспансия (увеличение)
нестабильных тринуклеотидных повторов
до 1000 раз (в норме их от 6 до 42 повторов).
Клиника:
важнейший клинический симптом —
олигофрения (слабоумие). Считается, что
болеют только мужчины. Однако есть
сведения, что около 30% женщин, гетерозиготных
носителей ломкого гена, также страдают
олигофренией. У мужчин, помимо олигофрении,
имеются и другие характерные признаки:
увеличенные в объеме яички, большие
уши, выпуклый лоб, выступающая челюсть,
речевые аномалии, среди которых широко
распространено заикание.
Диагностика:
кариотипирование — отрыв конечного
участка длинного плеча в Х-хромосоме,
что выявляется лишь при культивировании
лимфоцитов в условиях дефицита фолиевой
кислоты и внешне напоминает «спутник»
длинного плеча. Наиболее точный метод
– молекулярно генетическая диагностика.
Возможна пренатальная диагностика.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
Источник