Синдром джервелла и ланге нильсона

Синдром Жервелла и Ланге-Нильсена. Кардиоаудиторный или сурдокардиальный синдромJervell и Lange-Nielsen описали синдром глубокой врожденной нейросенсорной глухоты с патологией ЭКГ, характеризующейся удлинением интервала Q—Т, обморочными приступами и иногда внезапной необъяснимой смертью в детском возрасте. Впоследствии синдром был изучен многими авторами (Lcvine, Woodworth, Fraser, Froggatt, James, Jervell, Thingstad, Endsjo, Lisker, Finkelstein, Kallfelz, Sanchez Cascos et al., van Bruggen et al., Fauchier et al., Athanasiou, Weiner). Fraser, Froggatt и Murphy установили его частоту, составляющую от 3 до 4 случаев на 1 млн. рождений. Кроме того, они определили, что синдром может составлять до 1% среди наследственной глухоты. Однако Sanchez Cascos с сотр. нашли только один случай среди 511 глухонемых детей. Fay с соавт. обнаружили одного больного с этим синдромом среди 1100 глухих детей. Кроме того, 4 больных страдали обмороками без удлинения интервала Q—Т, а у 5 больных отмечалось удлинение интервала Q—Т без обмороков. Орган слуха. У всех больных отмечалась врожденная двусторонняя глубокая нейросенсорная глухота. Лабораторные данные. Изменения ЭКГ характеризовались удлинением интервала Q—Т с широкой T-волной, которая могла быть вертикальной, зазубренной, двухфазной или перевернутой. Степень удлинения интервала Q—Т варьировала как внутри одной семьи, так и между разными семьями, но почти всегда превышала 0,5 с (при максимуме 0,4 с В норме). Так как интервал Q—Т удлиняется в зависимости от скорости сердечных сокращений, существует простая формула для определения его величины (Ljting): Q—T= (RRX0,2) +0,18±0,04. В нескольких случаях была обнаружена легкая гипохромная анемия (Jervell, Langc-Nielsen, Fraser, Froggatt, James, Jervell et al., Lamy et al, Kallfelz). Патология. Описаны многочисленные результаты вскрытия. При макроскопическом и гистологическом исследовании сердца в большей части случаев патологии не обнаружили. Специальное исследование проводящей системы сердца показало выраженное сужение главной ветви артерии синусового узла, в результате чего развивался инфаркт узла (Friedmann et al.). Fraser, Froggatt и James обнаружили, что в сердце этих больных отсутствует нормальная гликогенсодержащая перинуклеарная зона в волокнах Пуркинье.
Изменения височной кости описаны Friedmann с соавт.. Наиболее уникальным изменением являлось накопление ПАСК-положительных глыбок гиалина в атрофичной сосудистой полоске. Отмечалась почти полная дегенерация кортиева органа с утратой чувствительных клеток. Покровная мембрана была сморщенной или втянутой, а рейснерова мембрана была сращена с основной мембраной, практически облитсрируя улитковый ход. Чувствительный эпителий утрикулюса и саккулюса был атрофичным, а гребешок дезорганизованным. Отмечалась умеренная утрата нервных клеток спирального узла. Наследственность. Синдром отчетливо наследуется по аутосомно-рецессивному типу. Часто встречаются кровнородственные браки между родителями (Fraser, Froggatt, Murphy, Lamy et al., Sanchez Cascos et al.). У гетерозигот может наблюдаться умеренное удлинение интервала Q—Т (Fraser, Froggatt и Murphy). Предполагают, что заболевание наследуется сцепленно с Rh-фактором (Friedmann, Fraser, Froggatt). Диагноз. Описаны некоторые изменения ЭКГ (такие, как удлинение интервала Q—Т) без глухоты (Johansson, Jorming, van der Straaten, Bruins, Singer et al.). В этих случаях также наблюдались внезапная смерть и/или приступы потери сознания. Однако патология в этих семьях наследовалась по доминантному типу. Jervell описал еще один доминантно наследующийся синдром множественных экстрасистол с приступами фибрилляции желудочков, но с нормальным интервалом Q—Т. Отмечается определенное сходство настоящего заболевания с изменениями, наблюдающимися при синдроме Рефсума. При обоих синдромах выражены нейросенсорная глухота, нарушения сердечной проводимости с удлинением интервала Q—Т и аномальной Т-волной и иногда наступает внезапная смерть. Однако при синдроме Рефсума глухота выявляется в зрелом возрасте и, кроме того, в сыворотке крови повышен уровень фитановой кислоты. Обморочные приступы при синдроме Ланге-Нильсена могут быть ошибочно диагностированы как эпилептические припадки. Однако ЭЭГ нормальна, в то время как ЭКГ резко изменена. Кроме того, после обморочных состояний у детей не развивается глубокого оглушения. Отмечено несколько атипичных случаев. Mathews с соавт. сообщили об аутосомно-доминантной форме с легкой высокочастотной глухотой. Глухота и сердечная патология могут наследоваться как независимые доминантные признаки. Furlancllo с сотр. и Athanasiou и Muller-Seydlitz также описали доминантную форму заболевания у взрослых с легким дефектом слуха. Предполагают также, что в этих семьях имелся синдром «LEOPARD». Читатель, конечно, помнит, что причиной удлинения интервала Q—Т могут служить: гипокалиемия, гипокальциемия, гипомагниемия и введение квинидина и фенотиазина. Прогноз. С возрастом отмечается незначительное прогрессирование дефекта слуха. У больных может наблюдаться различное число обморочных приступов. Примерно в половине случаев больные погибают к 15-летнему возрасту. Было выявлено несколько больных в возрасте старше 21 года. Выводы. Главными чертами этого синдрома являются: — Также рекомендуем «Ушная-зубная или отодентальная дисплазия. Липодистрофия лица, плеч и кисты костей с глухотой» Оглавление темы «Генетические болезни с глухотой»:
|
Источник
Джервелл и Ланге-Нильсена синдром ( JLNS ) представляет собой тип длинного интервала QT синдрома , связанного с тяжелой, двусторонней нейросенсорной потерей слуха . Длинный QT синдром приводит к сердечной мышце , чтобы занять больше времени , чем обычно , чтобы перезарядить между ударами. При отсутствии лечения нерегулярных сердцебиения, называемые аритмии , могут привести к обмороку , судорогам или внезапной смерти. Впервые он был описан Антон Джервелл и Фред Ланге-Нильсена в 1957 году.
Симптомы и признаки
Симптомы этого заболевания являются:
- Врожденная глухота
- синкопа
- Приступы
- Сердцебиение
- Внезапная сердечная смерть
Клинические признаки:
- Продолжительное QTc на ЭКГ
- Двусторонняя нейросенсорная глухота
генетика
Джервелл и Ланге-Нильсена синдром вызывается мутациями в KCNE1 и Kcnq1 генов. Белки , полученные этими двумя генами работают вместе , чтобы сформировать канал калия , который транспортирует положительно заряженный калия ионов из клеток , который называется медленной задержкой выпрямитель тока калия. Движение ионов калия через эти каналы имеет решающее значение для поддержания нормальных функций внутреннего уха и сердечной мышцы. JLNS является аутосомно — рецессивным расстройством означает , что две копии генетической мутации необходима для получения полного синдрома. Мутации в одних и тех же генах могут производить более мягкую Романо-Уорд форму длительного интервала QT синдрома , если только одна копии генетической мутации по наследству.
Около 90% случаев Джервелл и синдрома Ланге-Нильсена вызваны мутациями в Kcnq1 гена, что приводит к Джервелл и Ланге-Нильсена синдром 1 -го типа (JLNS1). KCNE1 мутации ответственны за оставшиеся 10% случаев, в результате чего Джервелл и Ланге-Нильсена синдром 2 -го типа (JLNS2). Мутации в этих генах изменяют обычную структуру и функцию калиевых каналов или предотвратить сборку нормальных каналов. Эти изменения нарушают поток ионов калия во внутреннем ухе и в сердечной мышце, что приводит к потере слуха и нерегулярного ритма сердца характеристики Джервелл и Ланге-Нильсена синдрома.
| Тип | OMIM | Ген | Заметки |
| JLNS1 | 192500 | KCNQ1 | Кодирует альфа-субъединицу медленного задержанного канала выпрямителя калия К V 7.1 , несущей калий ток I Ks . |
| JLNS2 | 176261 | KCNE1 | Зашифровывает норка, калиевые каналы β-субъединица. |
диагностика
Тесты, используемые может быть —
- ЭКГ
- холтеровское мониторирование
- Сердечный Мониторинг событий
- Слуховые тесты
- Генетическое тестирование
- Электролит тесты
- Имплантируемый регистратор петли (ILR) Мониторинг
- Стресс-тестирование ЭКГ / беговая дорожка ЭКГ
лечение
JLNS пациентов с Kcnq1 мутации особенно подвержены патологическим удлинением интервала QT, который предрасполагает их к эпизодам пуантах де пуантах и внезапной сердечной смерти . В связи с этим, если пациент имел синкопальных или история сердца, имплантируемый дефибриллятор сердца должны быть использованы в дополнение к бета — блокаторов , таких как пропранолол .
эпидемиология
Джервеллы и синдром Ланге-Нильсен влияют , по оценкам , один в 166000 до 625000 детей, и отвечают за менее чем 10% от всех случаев длительного интервала QT синдрома. Он имеет значительно более высокий уровень в Норвегии и Швеции на до одного в 200.000.
Рекомендации
внешняя ссылка
Эта статья включает в себя публичный текст домена из американской Национальной медицинской библиотеки
- запись GeneReview / NIH / UW на Джервелл и Lange-Nielsen синдром
Источник
Медицинская статья опубликована в рубрике: Кардиология, ЛОР, СИНДРОМЫ | Декабрь 9th, 2013
В 1957 г. Жервелл совместно с Ланге Нильсеном описали синдром, характеризующийся постоянным сочетанием врожденной глухоты, с приступами синкопы и характерными электрокардио графическими изменениями.
Синдром Жервелла Ланге Нильсена — генетическая аномалия с семейным характером, поражающая оба пола.
Этиопатогенез синдрома Жервелла Ланге Нильсена.
Не существует предположения, которое объяснило бы одновременное нарушение слуха и миокарда. Предположения тех, кто придерживается единого мнения, объясняет аномалию сердца гипокалиемией, а глухоту — нарушением кровоснабжения всего слухового аппарата.
Удлинение интервала Q — Т обусловлено нарушением реполяризации (с неуточненной этиологией), с переменчивой интенсивностью от одного дня к другому, определяя такое же переменчивое изменение интервала Q — Т.
Сердечное синкопе объясняется, как приступ типа Адамса-Стокса, или как приступ венечной ишемии. Смерть, наступившая при этих синкопах, может быть вызвана бесконечным удлинением интервала Q—Т, полной предсердно-желудочковой диссоциацией или фибрилляцией желудочков. Следует подчеркнуть, что синкопе появляются все реже и реже, по мере развития ребенка.
Синдром встречается, как у взрослых, так и у детей; по Fraser, представляет приблизительно 1% всех случаев врожденной глухонемоты. Наследственная передача происходит рецессивным образом, вероятно, как результат палеотропического эффекта аномального гена у гомозигот с полным клиническим проявлением; у гетерозигот синдром проявляется только удлинением интервала Q—T.
Симптоматология синдрома Жервелла Ланге Нильсена.
Врожденная глухота — всегда присутствует; она двусторонняя, обусловливая глухонемоту маленького ребенка. Глухота перцепторного типа, не затрагивает низкие звуки.
Приступы синкопе — повторные, вызываемые обыкновенно волнениями, холодом или даже без видимой причины, продолжаются несколько секунд и внезапно прекращаются.
Во время синкопы у больных отмечается:
- бледность;
- тахикардия;
- полипное;
- больной стонет, теряет или нет сознание;
- иногда появляются судороги.
Несколько минут после прекращения синкопе, у больного затуманенное сознание, затем постепенно приходит в нормальное состояние.
Диагностика синдрома Жервелла Ланге Нильсена.
Электрокардиографические характерные изменения заключаются в:
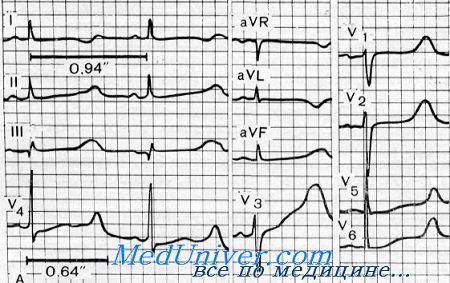
— патологическом удлинении интервала Q—Т (представляя постоянные и значительные электрокардиографические изменения). В сравнении с нормальным пределом этого интервала (0,31 секунды) величины, встречающиеся у больных, всегда намного выше (в среднем они колеблются между 0,40 и 0,55 секунд при сердечном ритме выше 90/мин).
Фонокардиограмма показывает, что длительность электрической систолы всегда больше механической:
- изменения волны Т очень разнообразна: отрицательная волна, положительная и двухфазная;
- комплекс QRS и интервал Р — Q — всегда нормальные.
Непостоянно добавляется и гипохромная анемия, умеренной интенсивности.
Течение и прогноз синдрома Жервелла Ланге Нильсена.
Течение, как и дальнейший прогноз, неблагоприятные. Смерть ребенка может произойти во время приступа синкопе, тем более, что часто приступ синкопе диагностируется, как приступ эпилепсии и лечат его, как таковой, лишая, таким образом, больного соответствующей экстренной терапии.
Лечение синдрома Жервелла Ланге Нильсена
а) Лечебное:
- экстренное назначение (если диагноз был уточнен) дигиталиса в 1% растворе, 5—6 капель в день, 5 дней в неделю (с еженедельным 2-х дневным перерывом во избежание накопления дигиталиса в миокарде), в течение 3—4 недель. Применением дигиталиса можно избежать смерти во время синкоп, укорачивая и нормализируя интервал Q — R, в отличие от хинидина, который способствует его удлинению (отсюда вытекает абсолютное противопоказание хинидина при приступах синкопе);
- хлористый калий в дозе от 0,5—1 г/день перорально;
- успокаивающие (диазепам).
б) Профилактическое:
- электрокардиографическое исследование всех новорожденных и грудных детей, страдающих судорогами и состоянием синкопе;
- внимание при дифференциальном диагнозе приступа синкопе, который ошибочно может быть принят за приступ эпилепсии; от своевременного и правильного диагноза зависит жизнь больного.
Источник