Синдром дисомии у хромосом картинки

- Авторы
- Резюме
- Файлы
- Ключевые слова
- Литература
Кравец В.С.
1, 2, 3
Ворсанова С.Г.
1, 2, 3
Юров И.Ю.
2, 1, 4
Колотий А.Д.
1, 2
Боченков С.В.
1
Гордеева М.Л.
1
Юров Ю.Б.
1, 2, 3
1 Обособленное структурное подразделение «Научно-исследовательский клинический институт педиатрии имени академика Ю.Е. Вельтищева» ФГБОУ ВО РНИМУ им. Н.И. Пирогова Минздрава России
2 ФГБНУ Научный центр психического здоровья
3 ФГБОУ ВО «Московский государственный психолого-педагогический университет»
4 ФГБОУ ДПО «Российская медицинская академия последипломного образования» Минздрава России
В статье рассматривается редкий случай задержки психоречевого развития, признаков аутизма и микроаномалий у мальчика 3 лет с дисомией хромосомы Y, рождённого в кровнородственном браке. Отмечается, что клинические признаки, обнаруженные у пробанда, не совсем совпадают с классическими симптомами синдрома дисомии хромосомы Y. Подчеркивается необходимость применения молекулярно-цитогенетических технологий, таких как флюоресцентная in situ гибридизация (FISH) и молекулярное кариотипирование (серийная сравнительная геномная гибридизация или arrayCGH) с целью выявления возможных геномных аномалий, не обнаруживаемых стандартным (классическим) цитогенетическим методом, способных повлиять на клиническую картину, отмеченную у данного пациента. Обсуждается возможное влияние на симптомокомплекс пробанда сочетания дисомии хромосомы Y и инбридинга (коэффициент инбридинга 1/32) в данной семье.
дисомия хромосомы y
задержка психоречевого развития
аутизм
инбридинг
FISH
1. Ворсанова С.Г., Шаронин В.О., Курило Л.Ф. Аномалии половых хромосом при нарушении репродуктивной функции у мужчин : обзор литературы // Пробл. репродукции. – 1998. – № 2. – С. 12-21.
2. Ворсанова С.Г., Юров Ю.Б., Чернышов В.Н. Медицинская генетика. – М. : Медпрактика, 2006. – 300 с.
3. Ворсанова С.Г., Воинова В.Ю., Юров И.Ю., Куринная О.С., Демидова И.А., Юров Ю.Б. Цитогенетические, молекулярно-цитогенетические и клинико-генеалогические исследования матерей детей с аутизмом: поиск семейных генетических маркеров аутистических расстройств // Журнал неврологии и психиатрии им. C.С. Kорсакова. — 2009. — Т. 109, № 6. — С. 54-64.
4. Ворсанова С.Г., Юров И.Ю., Куринная О.С., Воинова В.Ю., Юров Ю.Б. Геномные аномалии у детей с умственной отсталостью и аутизмом: использование технологии сравнительной геномной гибридизации на хромосомах in situ (HR CGH) и молекулярного кариотипирования на ДНК-микроматрицах (arrayCGH) // Журнал неврологии и психиатрии им. C.С. Корсакова. — 2013. — Т. 113, № 8. — С. 46-49.
5. Ворсанова С.Г., Юров И.Ю., Демидова И.А., Кравец В.С., Юров Ю.Б. Цитогенетика и молекулярная цитогенетика аутизма. — М. : Издательский дом Академии Естествознания, 2016. – 144 с.
6. Фогель Ф., Мотульски А. Генетика человека. — М. : Мир, 1989. – Т. 2. – С. 341.
7. Юров И.Ю., Ворсанова С.Г., Юров Ю.Б. Cовременные достижения в молекулярно-цитогенетической диагностике наследственных болезней (лекция) // Клиническая лабораторная диагностика. — 2005. — № 11. — С. 21-29.
8. Юров И.Ю., Ворсанова С.Г., Юров Ю.Б. Геномные и хромосомные болезни центральной нервной системы: молекулярные и цитогенетические аспекты. — М. : Медпрактика, 2014. – 384 с.
9. Юров И.Ю., Ворсанова С.Г., Зеленова М.А., Васин К.С., Юров Ю.Б. Биоинформатическая технология оценки функциональных последствий геномных вариаций // Фундаментальные исследования. – 2015. — № 2-19. – С. 4209-4214.
10. Юров Ю.Б., Ворсанова С.Г. Молекулярно-цитогенетические исследования хромосомных аномалий и нарушений при нервно-психических заболеваниях: поиск биологических маркеров для диагностики // Вестник РАМН. — 2001. — № 7. — С. 26-31.
11. Iourov I.Y., Vorsanova S.G., Yurov Y.B. Single cell genomics of the brain: focus on neuronal diversity and neuropsychiatric diseases // Current Genomics. — 2012. — Vol. 13. – N 6. — Р. 477-488.
12. Iourov I.Y., Vorsanova S.G., Korostelev S.A., Zelenova M.A., Yurov Y.B. Long contiguous stretches of homozygosity spanning shortly the imprinted loci are associated with intellectual disability, autism and/or epilepsy // Mol Cytogen. – 2015. – Vol. 8. – № 1. – 8 р.
13. Milazzo J.P., Rives N., Mousset-Simeon N., Mace B. Chromosome constitution and apoptosis of immature germ cells present in sperm of two 47,XYY infertile males // Hum. Reprod. – 2006. – 21. – P. 1749–1758.
14. Robinson D.O., Jacobs P.A. The origin of the extra Y chromosome in males with a 47, XYY karyotype // Hum. Mol. Genet. – 1999. – N 8. – P. 2205–2209.
15. Schinzel A. Catalogue of unbalanced chromosome aberrations in man. — Berlin, New York, de Gruyter, 2001. – 966 с.
16. Shah K., Sivapalan G., Gibbons N., Tempest H., Griffin D.K. The genetic basis of infertility // Reproduction. – 2003. – N 126. – P. 13-25.
17. Soloviev I.V., Yurov Y.B., Vorsanova S.G., Malet P. Microvawe acnivation of fluorescence in situ hybridization: a novel method for rapid chromosome detection and analysis // Focus. – 1994. – N 16 (4). – P. 115-116.
18. Soloviev I.V., Yurov Y.B., Vorsanova S.G., Fayet F., Roizes G., Malet P. Prenatal diagnosis of trisomy 21 using interphase fluorescence in situ hybridization of postreplicated cells with site-specific cosmid contig probes // Prenatal diagn. – 1995. – № 15. – Р. 237-238.
19. Yurov Y.B., Laurent A.-M., Marcais B., Vorsanova S.G., Roizes G. Analysis of pericentromeric chromosome 21 specific YAC clones by FISH: identification of new markers for molecular cytogenetic application // Hum. Genet. – 1995. – № 95. – Р. 287-293.
20. Yurov Y.B., Vorsanova S.G., Soloviev I.V., Demidova I.A., Alexandrov I.A., Sharonin V.O., Beresheva A.K. Original collection of DNA probes for preimplantational, fetal prenatal and postnatal diagnosis of chromosomal analysis by FISH / (eds): Macek M. Sr., Bianchi D., Cuckle H. Early prenatal diagnosis, fetal cells and DNA in mother, present state and perspectives. – Prague, 2002. — P. 275-283.
Среди обширного спектра хромосомных и геномных аномалий, обнаруживаемых у детей с задержкой психоречевого и полового развития, видное место занимают различные численные и структурные аномалии половых хромосом (гоносом). Наиболее изучены и часто встречаются синдромы Шерешевского-Тернера (кариотипы — 45,Х; 46,X,i(Xq); 45,X/46,XХ и другие), Клайнфельтера (кариотипы — 47,XXY; 48,XXXY и другие), трисомии хромосомы Х (кариотип — 47,ХХХ) и дисомии хромосомы Y (кариотип — 47,XYY) [2; 15]. Наблюдаются как регулярные, так и мозаичные формы этих синдромов; возможен тканевой мозаицизм. Клинические признаки пациентов сильно варьируют от почти полного их отсутствия, особенно в случаях мозаицизма с малой долей аномального клона, до выраженной умственной отсталости, пороков и микроаномалий развития, нарушения репродуктивных функций и других симптомов. Диагностика подобных случаев нередко, особенно при мозаицизме и структурных перестройках, требует применения таких молекулярно-цитогенетических методов исследований, как флюоресцентная гибридизация in situ (FISH) с хромосомоспецифичными и сайтспецифичными ДНК зондами [10], метафазная сравнительная геномная гибридизация, а также серийная сравнительная геномная гибридизация на ДНК-микроматрицах (array CGH), что позволяет уточнить генетический диагноз и проводить корректное медико-генетическое консультирование семей [3; 4; 7; 11].
Частота встречаемости синдрома дисомии хромосомы Y – 1-1,5:1000 новорождённых [1; 2; 14], а среди мужчин с психическими отклонениями пациенты с дисомией хромосомы Y встречаются в 0,45-15% случаев [15]. В основном дисомия хромосомы Y возникает в результате неправильного расхождения хромосом в отцовском мейозе II [16]. По данным литературы, при синдроме дисомии хромосомы Y отсутствуют обязательные признаки, характерные для гоносомных синдромов, но среди необязательных признаков часто отмечают высокий рост, умственную отсталость различной степени тяжести, нарушение половой дифференцировки (крипторхизм, гипогонадизм, дисплазия гениталий); у примерно 30% мужчин с этим синдромом отмечено нарушение репродуктивной функции [13], агрессивное, иногда асоциальное поведение, психопатические черты характера (импульсивность, отсутствие сильных привязанностей, плохое владение собой по поводу примитивных эмоций), черты аутизма; у некоторых больных выявляют шизофрению, депрессивные психозы, тяжёлые формы психопатии и эпилепсии. Среди других аномалий отмечают макроцефалию, прогнатизм, выступающие надбровные дуги, высокое нёбо, гипертрофию языка, увеличение конечностей. Показано также, что большинство опубликованных случаев данного синдрома обнаруживают во взрослых и детских психиатрических лечебницах и лечебно-профилактических учреждениях для содержания социально опасных пациентов, а также в тюрьмах [2; 5]. В целом для синдрома дисомии хромосомы Y характерно разнообразие клинических проявлений, что представляет большой интерес для клинических генетиков и заставляет тщательно исследовать каждый такой случай с целью накопления данных и установления корреляции «фенотип-генотип». Мы представляем результаты обследования ребенка мужского пола с дисомией хромосомы Y, имеющего клинические проявления, не совсем характерные для этого синдрома.
Материалы и методы
В работе обследовался мальчик в возрасте 3 лет. Культура лимфоцитов периферической крови, приготовление препаратов, дифференциальное окрашивание хромосом по длине и анализ кариотипа проводились по стандартным методикам (GTG- и CBG-окрашивание хромосом по длине) [2]. Кроме классического цитогенетического анализа, также было проведено молекулярно-цитогенетическое исследование – флюоресцентная in situ гибридизация (FISH) с центромерными хромосомоспецифичными ДНК-зондами на хромосомы Х и Y из оригинальной коллекции лаборатории [10; 18; 20]. Гибридизация и анализ препаратов проводились по стандартным протоколам [17; 19].
Результаты
В клинику института для обследования поступил ребёнок 3 лет, родившийся от 2-й беременности, 2-х физиологических родов с массой тела 3750 г, длиной — 54 см. От 1-й беременности родился здоровый мальчик, ему 5 лет. Причиной обращения послужили следующие клинические признаки: задержка психоречевого развития и аномалии поведения. При осмотре ребёнка отмечены такие симптомы, как ограниченность понимания обращённой речи, несформированность навыков опрятности и самообслуживания, стереотипные действия с предметами неигрового назначения, резко отрицательная реакция на запреты и ограничения в виде падения на пол и ударов головой об пол, а также агрессии к окружающим (укусы и т.д.), ходьба на цыпочках. Кроме того, у пациента обнаружены хронические кататоно-аффективные расстройства, аллергические реакции на пыль, плесень и шоколад, дисфункция желчного тракта, плоско-вальгусная деформация стоп, пролапс митрального клапана. Из родословной (рис. 1) видно, что прапрадеды пробанда по отцовской и материнской линиям являются родными братьями, и таким образом, родители пробанда – троюродные сибсы. В родословной, со слов родителей, не выявлено случаев умственной отсталости или пороков развития, у деда по отцовской линии обнаружен поллиноз, у деда по материнской линии – ишемическая болезнь сердца (ИБС).
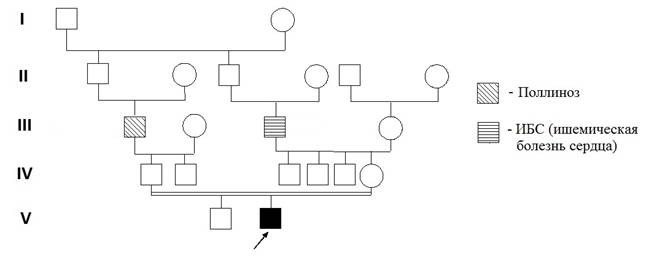
Рис. 1. Родословная пациента с синдромом дисомии хромосомы Y
На основании клинических признаков принято решение о проведении цитогенетического исследования. Из анализа кариотипа (рис. 2) видно, что у пробанда выявлены две хромосомы Y. В результате проведённых цитогенетических исследований кариотип пробанда (GTG- и CBG-окрашивания хромосом по длине) — 47,XYY. Других численных или структурных аномалий хромосом, обнаружимых стандартными цитогенетическими методами, у данного пациента не выявлено.
Для исключения мозаицизма проведено FISH исследование (рис. 3), при котором не обнаружено предполагавшегося хромосомного мозаицизма гоносом, и дисомию хромосомы Y следует считать регулярной и возникшей de novo: 47,XYY. ish (DXZ1x1;DYZ3x2) [100]. nuc ish (DXZ1x1;DYZ3x2) [1000]. Таким образом, после проведения цитогенетического и молекулярно-цитогенетического исследований ребёнку поставлен диагноз – синдром дисомии хромосомы Y.
На основании результатов цитогенетических и молекулярно-цитогенетических исследований с учётом родства родителей принято решение о дополнительном проведении пробанду молекулярно-цитогенетического исследования – серийной сравнительной геномной гибридизации (молекулярного кариотипирования или array CGH) [4; 8; 11]. Проведение молекулярного кариотипирования данному пациенту позволит выявить возможные аномалии генома, а также установить участки потери гетерозиготности (loss of heterozygosity, LOH), которые в данном случае ввиду родства родителей (троюродные сибсы) могут быть обширны и способны значительно (негативно) повлиять на клинические проявления у пробанда [12].
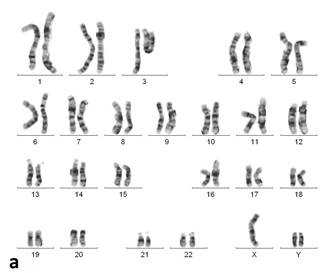

Рис. 2. Кариотип пробанда после проведения GTG- (а) и CBG- (б) окрашивания (47,XYY)
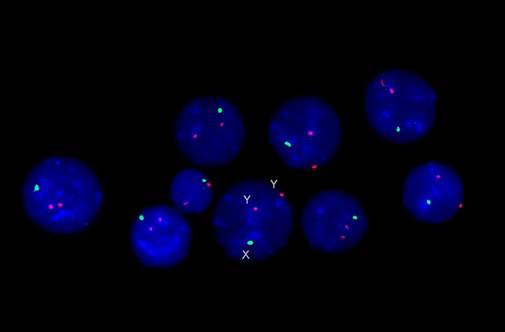
Рис. 3. Результаты FISH-исследования с центромерными ДНК-зондами на хромосомы Х и Y (зеленые сигналы – хромосома Х, красные – хромосома Y)
На рисунке представлены интерфазные ядра лимфоцитов периферической крови, в каждом из которых по две хромоcомы Y.
Обсуждение
В достаточно широком спектре различных численных и структурных аномалий половых хромосом (гоносом) дисомия хромосомы Y занимает особое место благодаря исключительной вариабельности клинических признаков данного синдрома: от почти полного отсутствия каких-либо патологических проявлений до умственной отсталости тяжёлой степени; бесплодие наблюдается лишь в 30% пациентов, и прежде всего это связано с нарушением сперматогенеза. Причины клинического полиморфизма при этом синдроме не всегда ясны и могут быть связаны с иными хромосомными (геномными) микроаномалиями, не обнаружимыми стандартными методиками классического кариотипирования [4; 7].
Данный случай необычен тем, что у пациента с регулярной дисомией хромосомы Y отмечены выраженные черты аутизма, что при данной патологии наблюдается нечасто [4]; вероятно, на картину заболевания повлияло и то, что ребёнок рождён в кровнородственном браке (родители – троюродные сибсы и принадлежат к одному и тому же этносу (аварцы) Северного Кавказа, где кровнородственные браки у некоторых народов нередки); поэтому у пробанда возможно наличие обширных участков потери гетерозиготности (LOH) более чем в 3% генома (коэффициент инбридинга 1/32) [6; 12]. Именно молекулярное кариотипирование и биоинформатический анализ in silico позволит выявить данные участки, что при их наличии может привести и к возникновению различных наследственных аномалий [8; 9]. С учётом всех данных этой семье рекомендовано медико-генетическое консультирование с возможной пренатальной диагностикой при повторном деторождении.
Заключение
В данной статье мы рассматриваем необычный случай синдрома дисомии хромосомы Y у мальчика, рождённого в кровнородственном браке. Изучение влияния на клинические проявления у пробанда как регулярной формы дисомии хромосомы Y, так и кровнородственного брака весьма важно для понимания вариабельности клинических проявлений синдрома дисомии хромосомы Y. Проведение array CGH (молекулярного кариотипирования на ДНК-микроматрицах) с последующим биоинформатическим анализом позволит выявить возможные дополнительные микроаномалии генома, не обнаруженные классическими цитогенетическими методами, а также участки потери гетерозиготности (LOH), которые тоже могут влиять на фенотип [8; 9].
Накопление данных о подобных случаях позволит более эффективно устанавливать корреляцию «фенотип-генотип», корректно описывать особенности фенотипа при различных хромосомных синдромах и проводить медико-генетическое консультирование семей с ребёнком, рождённым в кровнородственном браке.
Исследование выполнено при финансовой поддержке Российского научного фонда (проект № 14-15-00411).
Библиографическая ссылка
Кравец В.С., Ворсанова С.Г., Юров И.Ю., Колотий А.Д., Боченков С.В., Гордеева М.Л., Юров Ю.Б. ДИСОМИЯ ХРОМОСОМЫ Y У МАЛЬЧИКА С УМСТВЕННОЙ ОТСТАЛОСТЬЮ, АУТИЗМОМ И МИКРОАНОАНОМАЛИЯМИ РАЗВИТИЯ, РОЖДЁННОГО В КРОВНОРОДСТВЕННОМ БРАКЕ // Современные проблемы науки и образования. – 2017. – № 2.;
URL: https://science-education.ru/ru/article/view?id=26182 (дата обращения: 30.07.2020).
Предлагаем вашему вниманию журналы, издающиеся в издательстве «Академия Естествознания»
(Высокий импакт-фактор РИНЦ, тематика журналов охватывает все научные направления)
Источник
При такой генетической аномалии особь унаследует обе пары хромосом от одного родителя. Описаны различные варианты комбинаций такого наследования, например, передача двух одинаковых или двух разных хромосом. У человека при материнской однородительской дисомии чаще всего передаются 2, 7, 14 и 15 хромосомы, а при отцовской — 6, 11, 15, и 20 хромосомы.
Подобные дефекты могут приводить к серьезной патологии внутриутробного развития, последующему нарушению роста и поведения пациента и специфическим заболеваниям. Существует гипотеза, что однородительская дисомия развивается в результате трисомии плода на ранних сроках беременности. Так как данная генетическая аномалия является высоколетальной, плод сможет выжить, только потеряв лишнюю хромосому. То есть ученые считают, что однородительская дисомия — это механизм выживания плода за счет вытеснения нежизнеспособной трисомичной клеточной линии.
Полученные в результате серьезные нарушения могут затрагивать различные органы и системы. Среди наиболее актуальных заболеваний, которые развиваются вследствие однородительской дисомии, отмечаются муковисцидоз, синдром Прадера-Вилли и синдром Ангельмана.

Муковисцидоз
Главной причиной является мутация в гене CFTR. Однако в литературе описаны случаи развития заболевания в результате однородительской дисомии, в частности, когда оба мутировавших аллеля были унаследованы от одного родителя.
Проявления заболевания зависят от конкретной формы муковисцидоза. Это может быть поражение дыхательного аппарата, кишечника, повышенное отложение соли на коже, симптом барабанных палочек и др. Чтобы узнать, явилось ли заболевание следствием однородительской дисомии или других причин, пациент и его родители проходят генетическое тестирование.
Лечение муковисцидоза включает в себя пожизненную симптоматическую терапию.
Синдром Прадера-Вилли
Примерно у одной трети всех больных синдромом Прадера-Вилли выявляется однородительская дисомия по 15-й материнской хромосоме. Заболевание дает о себе знать уже в первые годы жизни ребенка. Оно характеризуется задержкой в росте и умственном развитии, нарушением строения половых желез, сниженным мышечным тонусом, ожирением, гинекомастией и др.
Синдром Прадера-Вилли иногда путают с синдромом Дауна, однако для опытного специалиста не составит труда отличить одно заболевание от другого. Чтобы поставить точку в этом вопросе, назначается исследование кариотипа ребенка и родителей, которое может подтвердить или опровергнуть, что болезнь развилась вследствие однородительской дисомии.
Лечение синдрома Прадера-Вилли симптоматическое. Оно включает в себя массаж, работу с логопедом, гормональную терапию и другие методы.
Синдром Ангельмана
Еще одним частым заболеванием, которое развивается вследствие однородительской дисомии, является синдром Ангельмана. Он проявляется в виде нарушений психического развития, непроизвольными движениями верхних и нижних конечностей, беспричинным смехом. Из-за таких специфичных симптомов синдром Ангельмана еще называют болезнью «счастливой куклы».
Данная патология развивается вследствие однородительской дисомии по 15-й отцовской хромосоме, кроме того, одной из причин её развития ученые называют мутацию в гене UBE3A. Чтобы установить точную причину синдрома Ангельмана, назначается генетическое тестирование.
Эффективного лечения заболевания, как и других видов патологии, связанных с однородительской дисомией, не разработано. Пациентам назначается симптоматическая терапия, которая помогает улучшить качество жизни и позволяет лучше адаптироваться в обществе.
Источник