Синдром ди джорджи мкб 10

Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Лечение
- Прогноз
- Профилактика
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Синдром Ди Джорджи.
Ребенок с синдромом Ди Джорджи
Описание
Синдром Ди Джорджи. Генетическое заболевание, относящееся к группе первичных иммунодефицитов и, наряду с ослаблением иммунитета, характеризующееся многочисленными пороками развития. Симптомами этого состояния являются частые бактериальные инфекции со склонностью к тяжелому течению, врожденные пороки сердца, аномалии развития лица и другие нарушения. Диагностика синдрома Ди Джорджи основывается на исследовании сердца, щитовидных и паращитовидных желез, изучении иммунологического статуса и данных молекулярно-генетических анализов. Лечение только симптоматическое, включает хирургическую коррекцию пороков сердца и аномалий лица, заместительную иммунологическую терапию, борьбу с бактериальными и грибковыми инфекциями.
Дополнительные факты
Синдром Ди Джорджи (гипоплазия тимуса и паращитовидных желез, велокардиофациальный синдром) – генетическое заболевание, обусловленное нарушением эмбрионального развития третьего и четвертого фарингеальных мешков. Впервые это состояние было описано в 1965 году американским педиатром Анджело Ди Джорджи, который классифицировал его как врожденную аплазию тимуса и паращитовидных желез. Дальнейшие исследования в области генетики помогли определить, что нарушения при этом заболевании выходят далеко за рамки первичного иммунодефицита. Это дало основание для появления другого названия синдрома Ди-Джорджи. С учетом наиболее часто поражаемых органов (небо, сердце, лицо) некоторые специалисты именуют данную патологию велокардиофациальным синдромом. Ряд современных исследователей разграничивают эти два состояния и считают, что «истинный» велокардиофациальный синдром не сопровождается выраженными иммунологическими нарушениями. Встречаемость синдрома Ди Джорджи составляет 1:3 000-20 000 – такое значительное расхождение данных обусловлено тем, что достоверная и четкая граница между этим заболеванием и велокардиофациальным синдромом до сих пор не установлена. Поэтому один и тот же больной, по мнению разных специалистов, может иметь либо первичный иммунодефицит, сопровождающийся сопутствующими нарушениями, либо многочисленнее пороки развития на фоне снижения иммунитета.
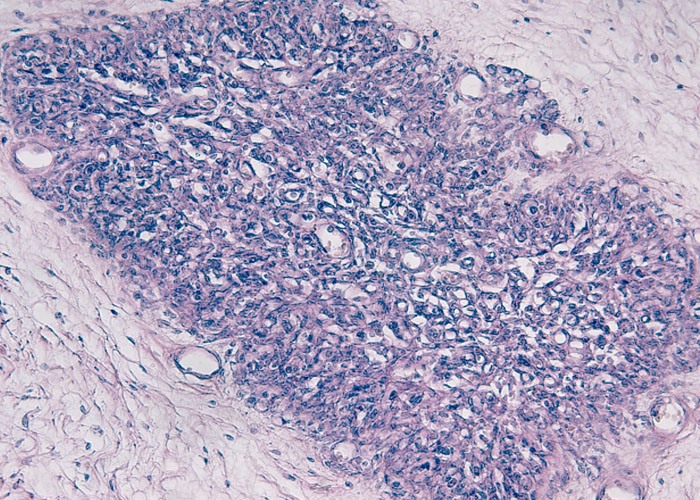
Гипоплазия тимуса при синдроме Ди Джорджи
Причины
Генетическая природа синдрома Ди Джорджи заключается в повреждении центральной части длинного плеча 22-й хромосомы, где предположительно располагаются гены, кодирующие ряд важных факторов транскрипции. Удалось идентифицировать один из этих генов – TBX1, продуктом его экспрессии является белок под названием T-box Он относится к семейству протеинов, контролирующих процессы эмбриогенеза. Доказательством взаимосвязи синдрома Ди Джорджи и TBX1 является тот факт, что незначительный процент больных не имеет выраженных повреждений 22-й хромосомы, присутствуют только мутации в этом гене. Также высказываются предположения о роли делеций иных хромосом в развитии данного заболевания. Так, аналогичные синдрому Ди Джорджи проявления выявлялись при наличии повреждений 10-й, 17-й и 18-й хромосом.
В большинстве случаев синдрома Ди Джорджи делеция 22-й хромосомы захватывает порядка 2-3 миллионов пар оснований. Чаще всего данный генетический дефект возникает спонтанно во время формирования мужских или женских половых клеток – то есть, носит герминативный характер. Лишь десятая часть всех случаев заболевания представляет собой семейную форму с аутосомно-доминантным характером наследования. Патогенез синдрома Ди Джорджи сводится к нарушению формирования особых эмбриональных образований – фарингеальных мешков (главным образом, 3-го и 4-го), которые являются предшественниками ряда тканей и органов. Главным образом, они отвечают за формирование неба, паращитовидных желез, тимуса, сосудов средостения и сердца, поэтому при синдроме Ди Джорджи возникают пороки развития именно этих органов.
Симптомы
Многие проявления синдрома Ди Джорджи определяются сразу после рождения ребенка, отдельные пороки развития (например, сердца) можно выявить еще раньше – на профилактических ультразвуковых исследованиях. Чаще всего первыми обнаруживаются аномалии развития лица – расщепление неба, иногда в сочетании с «заячьей губой», прогнатия нижней челюсти. Зачастую младенцы с синдромом Ди Джорджи имеют небольшой рот, маленький нос с расширенной переносицей, деформированные или недоразвитые хрящи ушных раковин. При относительно легком течении заболевания все вышеперечисленные симптомы могут быть выражены довольно слабо, даже расщепление твердого неба может возникать только в задней его части и выявляться лишь при тщательном осмотре у отоларинголога.
В первые месяцы жизни больного синдромом Ди Джорджи на первый план выступают проявления врожденных пороков сердца – это может быть как тетрада Фалло, так и отдельные нарушения: дефект межжелудочковой перегородки, незаращение артериального протока и ряд других. Они сопровождаются цианозом, сердечно-сосудистой недостаточностью и при отсутствии квалифицированной медицинской помощи (в том числе и хирургической) могут приводить к ранней смерти больных. Другим распространенным нарушением у детей с синдромом Ди Джорджи считаются судороги и тетания, обусловленная гипоплазией паращитовидных желез и последующей гипокальциемией.
Лимфоцитопения. Судороги.
Диагностика
Для определения синдрома Ди Джорджи применяют метод физикального общего осмотра, кардиологические исследования (ЭхоКГ, электрокардиограмма), УЗИ щитовидной железы и тимуса, иммунологические пробы. Вспомогательную роль играет проведение общего и биохимического анализов крови, изучение анамнеза больного, генетические исследования. При осмотре больных синдромом Ди Джорджи могут определяться характерные для заболевания нарушения – расщепление твердого неба, аномалии строения лица, патологии ЛОР-органов. В анамнезе, как правило, выявляются частые эпизоды вирусных и грибковых инфекций, принимающих тяжелое течение, судороги, обусловленные гипокальциемией, нередко обнаруживается обширное кариозное поражение зубов.
На ультразвуковых исследованиях вилочковой железы отмечается значительное уменьшение массы или даже полное отсутствие органа (агенезия). ЭхоКГ и другие кардиологические методы диагностики выявляют многочисленные пороки сердца (например, дефект межжелудочковой перегородки) и сосудов средостения. Иммунологические исследования подтверждают значительное падение уровня Т-лимфоцитов. Это же явление наблюдается в периферической крови и нередко сочетается с уменьшением концентрации белков-иммуноглобулинов. Биохимическое изучение крови свидетельствует о снижении уровня кальция и гормонов паращитовидной железы. Врач-генетик может выполнить поиск делеций в 22-й хромосоме посредством флуоресцентной гибридизации ДНК или мультиплексной полимеразной цепной реакции.
Лечение
Специфического лечения синдрома Ди Джорджи на сегодняшний момент не существует, используют только паллиативные и симптоматические методики. Очень важно как можно раньше выявить врожденные пороки сердца и при необходимости произвести их хирургическую коррекцию, поскольку именно сердечно-сосудистые нарушения являются наиболее частой причиной неонатальной смерти при этом заболевании. Значительную опасность представляют собой судорожные приступы, обусловленные гипокальциемией, что требует своевременной коррекции электролитного баланса плазмы крови. Помощь хирургов при синдроме Ди Джорджи также может потребоваться для устранения пороков развития лица и неба.
Из-за выраженного иммунодефицита любые признаки бактериальной, вирусной или грибковой инфекции являются поводом для срочного применения соответствующих препаратов (антибиотиков, противовирусных и фунгицидных средств). Для улучшения иммунного статуса больного синдромом Ди Джорджи может производиться заместительное вливание иммуноглобулинов, полученных из донорской плазмы. В отдельных случаях осуществлялась пересадка вилочковой железы, которая стимулировала образование собственных Т-лимфоцитов – это способствовало улучшению качества жизни больных.
Прогноз
Прогноз синдрома Ди Джорджи большинством исследователей оценивается как неопределенный, так как данное заболевание характеризуется значительной вариабельностью симптомов. В тяжелых случаях имеется высокий риск ранней неонатальной смерти из-за сочетания сердечно-сосудистых и иммунологических нарушений.
Профилактика
Более доброкачественные формы синдрома Ди Джорджи требуют достаточно интенсивной паллиативной терапии, особенно важно уделять внимание лечению и профилактике вирусных и грибковых инфекций. Интеллектуальное развитие больных несколько замедлено, однако при правильной педагогической и психологической коррекции проявления задержки развития можно нивелировать. Из-за частого спонтанного характера мутаций профилактика синдрома Ди Джорджи не разработана.
Основные медуслуги по стандартам лечения | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Клиники для лечения с лучшими ценами
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Источник
Общие сведения
Синдром Ди Джорджи еще в некоторых источниках указывают как синдром Ди Георга. Впервые патология была описана в 1965 году американским педиатром и эндокринологом — Анджело Мария Ди Джорджи (фото). По международной классификации десятого пересмотра (МКБ-10) данному иммунодефициту был присвоен код D82.1 и указан как синдром дивертикула глотки, вилочковой железы: алимфоплазия, аплазия либо гипоплазия с иммунной недостаточностью.
")
Angelo DiGeorge(15.04.1921-11.10.2009)
Синдром Ди Джорджа как первичный иммунодефицит является редким врожденным заболеванием (1 случай на 4-6 тыс. человек) и относится к идиопатическому изолированному гипопаратиреозу – состоянию, для которого характерно снижение выработки паратгормонов и гипокальциемия. Для синдрома свойственно множество фенотипов поражения, осложнений и присоединения сопутствующих нарушений.
Патогенез
Для синдрома Ди Георга характерна аплазия или гипоплазия вилочковой железы (тимуса), а также агенезия или дисгенезия паращитовидных желез в результате нарушений закладки и эмбриональной дифференцировки 3-4 жаберных (глоточных) карманов. Это вызывает резкое снижение популяции Т-лимфоцитов, нарушение их дифференцировки и как следствие — иммунологическую недостаточность. У детей могут развиваться различные врождённые аномалии крупных сосудов, например, дефекты аорты, межжелудочковой перегородки или синий порок сердца.

Расположение паращитовидных и щитовидных желез
Классификация
В зависимости от спектра клинических проявлений и врожденных аномалий синдром Ди Георга может быть полным и частичным, к примеру, протекать как изолированная недостаточность паращитовидных желёз либо врождённое отсутствие паращитовидных (околощитовидных) желез, которые приводят к гипокальциемическим судорогам, наблюдающимся у новорожденных в виде неонатальной тетании. Нарушения закладки тимуса приводят к развитию различных инфекционных заболеваний и обычно вызваны иммунологической недостаточностью.
Достаточно часто болезнь протекает в менее тяжелой форме и является наиболее распространенной причиной умственной отсталости с малым количеством других проявлений, поэтому может быть формально не диагностирована.
Причины
У синдрома Ди Джорджи или врождённой аплазии тимуса и паращитовидной железы генетическая причина развития. Патология развивается в результате делеции центрального участка более длинного плеча двадцать второй хромосомы, поэтому еще обозначается как синдром 22q 11.2. Однако, в некоторых случаях аналогичная клиническая картина наблюдалась и после других хромосомных перестроек – транслокаций и микроделеций, развивающихся в таких хромосомах как ТВХ1, 10р13, 17р13, 18q21 и пр. Эти мутации спорадические в 90% и обычно происходят на этапе мейоза спермато— или овогенеза.
Для данного синдрома дисэмбриогенеза 3-4 жаберной дуги — фенотипа CATCH 22 характерно наследование по аутосомно-доминантному типу, которое наблюдается лишь в 5-10% случаев, но при этом не исключается возможность аутосомно-рецессивного типа наследования с разной экспрессивностью.
Симптомы врождённой аплазии тимуса и паращитовидных желёз (Синдром Ди Джорджи)
Синдром Ди Джорджи – это первичный иммунодефицит, вызванный врожденным отсутствием или наличием аномалий развития тимуса и паращитовидных желез, который отличается триадой клинических симптомов, включающей:
- различные врожденные пороки сердца, дефекты крупных сосудов, носа, рта, ушей, что в результате дает специфические черты лица (как на фото): гипертелоризм (увеличенное расстояние между глазами), микрогнатия (недоразвитие нижней челюсти), низкое расположение ушных раковин;
- первичный иммунодефицит – нарушен как клеточный, так и гуморальный ответ иммунитета в результате аплазии вилочковой железы и невозможности обеспечения нормального развития Т-клеток;
- прогрессирующая гипокальциемия (пониженная концентрация кальция в кровотоке) – вызвана гипопаратиреозом, сопровождается судорожным синдромом и развитием скелетных аномалий.

Ребенок со специфическими чертами, характерными для синдрома Ди Джорджи
Особенности синдрома могут широко варьироваться даже в пределах одной семьи и затрагивать различные системы органов. Сопутствующими проблемами становится:
- нарушение работы почек и их атрофия, развитие нефрокальциноза, гидронефроза и пр.;
- проблемы с пищеварением и перистальтикой ЖКТ;
- дефицит гормона роста;
- проблемы с речью;
- психические расстройства, в том числе шизофрения;
- потеря слуха (проводящая и нейросенсорная обычно вызвана черепно-лицевыми синдромами);
- нарушения координации, которые могут быть вызваны гипоплазией мозжечка;
- синюшность кожных покровов в результате плохого кровообращения;
- ревматоидный артрит;
- частые переломы костей;
- болезнь Грейвса;
- болезнь Паркинсона.
Анализы и диагностика
Для подтверждения диагноза синдром Ди Джорджа необходимо выявление агенезии или дисгенеза паращитовидных желёз, аплазии вилочковой железы, иммунологической недостаточности, черепно-лицевых дисморфий (микрогнатии, гипертелоризма, антимонголоидного разреза глаз, расщелины губы и нёба, деформированных и/или низко расположенных ушных раковин) и прочих типичных аномалий развития. При этом оказываются эффективными различные исследования, включая УЗИ, МРТ и пр., а также используются методы генетического тестирования, к примеру, FISH (метод флуоресцентной гибридизации in situ).
Наиболее яркими проявлениями является гипопаратериоз и молочница. Также при плановом обследовании выявляются дефекты аорты, тетрада Фалло, катаракта, паховая грыжа и тому подобное. При помощи серологических исследований можно выявить лимфоцитопению, гипокальциемию, гипоγ-глобулинемии. Благодаря иммунологическим методам удается обнаружить нарушения трансформации лимфоцитов и их функциональной активности, в 20% случаев наблюдается сниженное количество Т-клеток. После иммунологических исследований важно провести дифференциальный диагноз с прочими первичными иммунодефицитами, таким как: синдром Вискотта-Олдрича, Брутона.
Лечение
Так как недуг неизлечим больным обычно назначают симптоматическое и комплексное поддерживающее лечение. Основными приемами считается соблюдение диеты, стерильные условия, прием кальция, комплексов витамин, антибиотиков, противогрибковых, иммуномодулирующих, седативных препаратов и пр.
Доктора
Лекарства
В зависимости от особенностей течения и имеющихся проблем со здоровьем могут быть назначены:
- противовирусные, противогрибковые препараты, а также антибиотики широкого спектра действия для лечения инфекционных заболеваний;
- пожизненный прием витамина Д и кальция;
- заместительная терапия иммуноглобулином.
Процедуры и операции
Для наилучшего прогноза пациентов с синдромом Ди Георга необходимо раннее выявление всех аномалий развития и их устранение стандартными методами лечения:
- трансплантация эмбриональных тканей тимуса;
- кардиохирургия при врожденных пороках сердца.
Первичный иммунодефицит у детей
В результате клеточного иммунодефицита дети с синдромом Ди Джорджи очень подвержены действию инфекции вирусной, грибковой и некоторых разновидностей бактериальной природы. Чаще всего развивается затяжной и тяжелый ринит, пневмония, абсцесс, пиелонефрит, колит, сепсис и т.д. Если ребенку удается пережить пятилетний возраст, то может не обнаруживаться недостаточность Т-клеток, так как антиген-независимый этап созревания Т-клеток начинает происходить не в тимусе, а в многослойном плоском эпителии.
У новорожденных часто возникают судороги и проблемы с кормлением. Младенцы слабые, у них плохой аппетит. Нарушение перистальтики приводит к частым запором. В дальнейшем у деток наблюдается задержка роста и развития, возникают проблемы с обучением, а также когнитивные нарушения, например, приобретение аутистических черт поведения.
У детей с синдромом ДиДжорджа специфический профиль, выявляемый нейропсихологическими тестами. У них нормальный IQ и они способны проходить обучение в обычной школе, дома или заниматься в специальных классах, ведь очень часто у больных есть проблемы с речью (неразборчивость, ошибки артикуляции, сложности при приобретении словарного запаса).
Прогноз синдрома Ди Джорджи
К сожалению, не сегодняшний день, современная медицина оказывается бессильна перед данным генетическим заболеванием. Прогноз крайне неблагоприятный – дети умирают от различных инфекционных заболеваний или сердечной недостаточности, поэтому лучшим способом предупреждения развития врожденных патологий считается медико-генетическое консультирование и пренатальная диагностика наследственных болезней.
Список источников
- Стефани Д.В., Вельтищев Ю.Е. Иммунология и иммунопатология детского возраста.— М., 1996, -С. 27.
- Ивановская Т.Е., Цинзерлинг А.В. Патологическая анатоми.— М., 1976. -С. 136.
Источник