Синдром ди джорджи гипоплазия тимуса

Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 29 ноября 2019;
проверки требует 1 правка.
Синдром Ди Георга (синдром Ди Джорджа, синдром Ди Джорджи, синдром дисэмбриогенеза 3-4 жаберной дуги, врождённая аплазия тимуса и паращитовидных желёз, синдром 22q11.2, CATCH 22 phenotype[2]) — разновидность идиопатического изолированного гипопаратиреоза; редкое врождённое заболевание. Генетической причиной синдрома Ди Георга является делеция центрального участка длинного плеча хромосомы 22 (22q11.2) размером 1.5-3 млн.п.н. Однако известны случаи, когда при тех же клинических проявлениях имеет место делеция других хромосом — 10р13, 17р13, 18q21 и других. В большинстве случаев делеция происходит во время мейоза при спермато- или овогенезе. Только в 5-10 % случаев дефектная хромосома наследуется по аутосомно-доминантному типу[3]. Характеризуется агенезией или дисгенезом паращитовидных (околощитовидных) желёз, аплазией тимуса (вилочковой железы), приводящей к резкому снижению популяции Т-лимфоцитов и иммунологической недостаточности, врождёнными аномалиями крупных сосудов (дефекты аорты, тетрада Фалло)[4].
Этиология и патогенез[править | править код]
Патология 22-й хромосомы (22q11.2) наследуется по аутосомно-доминантному типу.
Наиболее вероятная причина развития клинической симптоматики при данном синдроме — несбалансированная транслокация, делеция или микроделеция 22-й хромосомы (22q11.2). Большинство случаев спорадические, обусловлены делециями 22q11[5].
Заболевание развивается в результате повреждения закладки 3-4 жаберных карманов, в результате которого нарушается закладка паращитовидных желёз и тимуса. Тип наследования до конца не установлен — некоторые авторы предполагают аутосомно-доминантный тип с различной экспрессивностью[6].
Клиническая картина[править | править код]
Клинически наиболее постоянными проявлениями заболевания является гипопаратиреоз и кандидомикоз, отмечаются аномалии развития носа, рта, ушей[4].
Заболевание характеризуется аплазией тимуса и связано с нарушениями развития тимуса в эмбриональном периоде. Тимусный эпителий не может обеспечить нормальное развитие Т-клеток. В результате у пациентов с данной формой иммунодефицита страдает как клеточный, так и гуморальный иммунный ответ. Дети с подобным иммунодефицитным заболеванием проявляют повышенную чувствительность к вирусным, грибковым и некоторым бактериальным инфекциям.
Возможно течение синдрома в виде изолированной недостаточности паращитовидных желёз или врождённого отсутствия околощитовидных (паращитовидных) желез — гипокальциемические судороги, начиная с периода новорожденности (тетания) и вилочковой железы (различные инфекционные заболевания как следствие иммунологической недостаточности)[6].
Диагностика[править | править код]
Основывается на выявлении типичных для синдрома аномалий развития[6]:
- агенезия или дисгенез паращитовидных желёз;
- аплазия вилочковой железы;
- иммунологическая недостаточность;
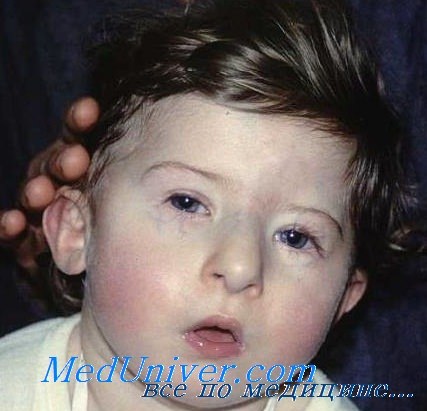
- черепно-лицевые дисморфии (микрогнатия, гипертелоризм, антимонголоидный разрез глаз, расщелины губы и нёба, деформированные и/или низко расположенные ушные раковины).
Наиболее яркие проявления — гипопаратиреоз и кандидомикоз. Возможно сочетание с дефектами аорты и тетрадой Фалло. Иногда — катаракта и паховые грыжи. В крови определяется лимфоцитопения, гипокальциемия, гипогамма-глобулинемия[6].
Прогноз[править | править код]
Обычно больные умирают в раннем возрасте от инфекционных заболеваний и сердечной недостаточности[4].
См. также[править | править код]
- DGCR2 — ассоциированный ген
- Гипокальциемический криз
- Тетания
Примечания[править | править код]
- ↑ Disease Ontology release 2019-05-13 — 2019-05-13 — 2019.
- ↑ Burn J. Closing time for CATCH22 (англ.) // J. Med. Genet. (англ.)русск. : journal. — 1999. — October (vol. 36, no. 10). — P. 737—738. — doi:10.1136/jmg.36.10.737. — PMID 10528851.
- ↑ Синдром Ди Джорджи. Центр Молекулярной Генетики при Медико-генетическом научном центре. Дата обращения 18 марта 2016.
- ↑ 1 2 3 Малая энциклопедия врача-эндокринолога / Под ред. А. С. Ефимова. — 1-е изд. — К.: Медкнига, ДСГ Лтд, Киев, 2007. — С. 312. — 360 с. — («Библиотечка практикующего врача»). — 5000 экз. — ISBN 966-7013-23-5.
- ↑ Эндокринология / Под ред. Н. Лавина. — 2-е изд. Пер. с англ. — М.: Практика, 1999. — С. 485, 483, 441—442, 66, 64.. — 1128 с. — 10 000 экз. — ISBN 5-89816-018-3.
- ↑ 1 2 3 4 Симптомы и синдромы в эндокринологии / Под ред. Ю. И. Караченцева. — 1-е изд. — Х.: ООО «С.А.М.», Харьков, 2006. — С. 67-68. — 227 с. — (Справочное пособие). — 1000 экз. — ISBN 978-966-8591-14-3.
Ссылки[править | править код]
- Медиафайлы по теме Синдром Ди Георга в Викискладе
Источник
Гипоплазия и аплазия тимуса. Синдром Ди Джорджи
При нарушении функции Т-лимфоцитов инфекционные и другие заболевания протекают, как правило, тяжелее, чем при недостаточности антител. Больные в таких случаях обычно погибают еще в грудном или раннем детском возрасте. Продукты поврежденных генов установлены лишь для некоторых первичных нарушений функции Т-лимфоцитов. Методом выбора при лечении этих больных в настоящее время является трансплантация тимуса или костного мозга от совместимых по HLA сибсов или гаплоидентичных (полусовместимых) родителей.
Гипоплазия или аплазия тимуса (вследствие нарушения его закладки на ранних стадиях эмбриогенеза) часто сопровождается дизморфией паращитовидных желез и других структур, формирующихся в то же время. У больных наблюдаются атрезия пищевода, расщепление нёбного язычка, врожденные пороки сердца и крупных сосудов (дефекты межпредсердной и межжелудочковой перегородки, правосторонняя дуга аорты и др.).
Типичные черты лица больных гипоплазией тимуса: укорочение губного желобка, гипертелоризм, антимонголоидный разрез глаз, микрогнатия, низко расположенные уши. Нередко первым указанием на данный синдром служат гипокальциемические судороги у новорожденных. Аналогичные черты лица и аномалии отходящих от сердца крупных сосудов наблюдаются при фетальном алкогольном синдроме.
Генетика и патогенез гипоплазии тимуса
Синдром Ди Джорджи встречается как у мальчиков, так и у девочек. Семейные случаи редки, и поэтому его не относят к наследственным заболеваниям. Однако более чем у 95% больных найдены микроделеции участков сегмента qll.2 хромосомы 22 (специфичный для синдрома Ди Джорджи участок ДНК). Эти делении, по-видимому, чаще передаются по материнской линии.
Их можно быстро обнаружить путем генотипирования с помощью ПЦР микросателлитных ДНК-маркеров, расположенных в соответствующей области. Аномалии крупных сосудов и делении участков длинного плеча хромосомы 22 объединяют синдром Ди Джорджи с велокардиофациальным и конотрункальным лицевым синдромом. Поэтому в настоящее время говорят о синдроме САТСН22 (Cardiac, Abnormal facies, Thymic hypoplasia, Cleft palate, Hypocalcemia — пороки сердца, аномалии строения лица, гипоплазия тимуса, расщепление нёба, гипокальциемия), включающем широкий спектр состояний, связанных с делециями 22q. При синдроме Ди Джорджи и велокардиофациальном синдроме найдены также делеции участков сегмента р13 хромосомы 10.
Концентрация иммуноглобулинов в сыворотке при гипоплазии тимуса обычно нормальная, но уровень IgA бывает сниженным, a IgE — повышенным. Абсолютное число лимфоцитов лишь ненамного ниже возрастной нормы. Число CD Т-лимфоцитов уменьшено в соответствии со степенью гипоплазии тимуса, и поэтому доля В-лимфоцитов оказывается повышенной. Реакция лимфоцитов на митогены зависит от степени недостаточности тимуса.
В тимусе, если он присутствует, обнаруживают тельца Гассаля, нормальную плотность тимоцитов и четкую границу между корковым и мозговым веществом. Лимфоидные фолликулы обычно сохранены, но в парааортальных лимфатических узлах и тимусзависимой области селезенки количество клеток обычно снижено.
Клинические проявления гипоплазии тимуса
Чаще имеет место не полная аплазия, а лишь гипоплазия тимуса и паращитовидных желез, называемая неполным синдромом Ди Джорджи. Такие дети растут нормально и не слишком страдают от инфекционных заболеваний. При полном синдроме Ди Джорджи, как и у больных с тяжелым комбинированным иммунодефицитом, повышена восприимчивость к условнопатогенной и оппортунистической флоре, включая грибы, вирусы и P. carinii, а при переливании необлученной крови часто развивается реакция «трансплантат против хозяина».
Лечение гипоплазии тимуса — синдрома Ди Джорджи
Иммунодефицит при полном синдроме Ди Джорджи корригируют пересадкой тканевой культуры тимуса (не обязательно от родственников) или нефракционированного костного мозга от HLA-идентичных сибсов.
— Также рекомендуем «Нарушения рецепторов CD3 Т-клеток и синтеза цитокинов»
Оглавление темы «Наследственные нарушения иммунитета»:
- Х-сцепленная агаммаглобулинемия. Брутоновская агаммаглобулинемия у детей
- Общая вариабельная гипогаммаглобулинемия (ОВГГГ) у детей. Причина и клиника
- Дефицит изолированный иммуноглобулина А — IgA у детей
- Гипогаммаглобулинемия у детей. Недостаток иммуноглобулинов G (IgG) у ребенка
- Х-сцепленный синдром гиперпродукции иммуноглобулина М (IgM) у мальчиков. Мутация CD40 — CD154
- Аутосомно-рецессивный синдром гиперпродукции иммуноглобулина М (IgM). Мутация гена AID
- Х-сцепленный лимфопролиферативный синдром мальчиков. Болезнь Дункана
- Лечение нарушений синтеза иммуноглобулинов. Терапия патологии В-лимфоцитов
- Гипоплазия и аплазия тимуса. Синдром Ди Джорджи
- Нарушения рецепторов CD3 Т-клеток и синтеза цитокинов
Источник

Синдром Ди Джорджи (синдром Ди Георга) — генетически детерминированная дисфункция иммунной системы с многочисленными аномалиями и морфофункциональными нарушениями в организме, возникающими в результате хромосомных мутаций. Первичный иммунодефицит сопровождается отсутствием или частичным недоразвитием тимуса, врожденными дефектами в структуре сердца и крупных сосудов, мальформацией лица. Синдром является разновидностью идиопатического изолированного гипопаратиреоза. Это наиболее распространенная форма патологии. “Полный” синдром Ди Джорджи проявляется поражением скелета, почек, глаз и встречается довольно редко. Такие больные нуждаются в консультации врачей разных специальностей.

ребенок с синдромом Ди Джорджи
В результате нарушения эмбрионального развития третьего и четвертого фарингеальных мешков возникает грубая аномалия тимуса. Этот орган располагается в средостении ребенка и обеспечивает выработку Т-лимфоцитов, отвечающих за формирование клеточного иммунитета. В вилочковой железе созревают, дифференцируются и иммунологически «обучаются» T-клетки иммунной системы. В период полового созревания орган претерпевает обратное развитие и стремительно уменьшается в размерах. Железистая ткань постепенно замещается жировой. Такие изменения происходят в организме здоровых людей.

Гипофункция вилочковой железы приводит к патологическому развитию клеток иммунной системы — Т-лимфоцитов, которые в норме помогают организму противостоять патогенным биологическим агентам. Неполноценное функционирование иммунокомпетентных клеток заканчивается снижением защитных сил организма. У пациентов с данным синдромом часто возникают тяжелые бактериальные инфекции. Снижение активности паращитовидных желез приводит к нарушению обмена фосфора и кальция с характерными клиническими проявлениями.
Впервые синдром описал детский врач из Америки Ди Джорджи в 1965 году. Он определил, что в основе синдрома лежит врожденное отсутствие вилочковой и паращитовидных желез. В дальнейшем ученые-генетики изучили механизм развития и основные проявления синдрома. В связи с поражением твердого неба, сердца и лица современные специалисты переименовали патологию в велокардиофациальный синдром. Но это неоднозначное мнение. В основе классического синдрома Ди Джорджи лежит первичный иммунодефицит. Велокардиофасциальный синдром проявляется многочисленными пороками развития на фоне снижения иммунной защиты.
Результаты исследования работы сердца, желез внутренней секреции, органов иммунной системы позволяют поставить правильный диагноз. Синдром Ди Джорджи — неизлечимое заболевание. Для улучшения общего состояния больных специалисты назначают симптоматическую и иммунозаместительную терапию, противомикотическое или антибактериальное лечение. Врожденные пороки сердца и мальформации лица устраняются оперативным путем.
Заболевание встречается одинаково часто как среди новорожденных мальчиков, так и среди девочек.
Этиология и патогенез
Синдром Ди Джорджи — генетический недуг, основанный на выпадении участка 22 хромосомы. Именно здесь локализуются гены, кодирующие ряд важных факторов, участвующих в переносе наследственной информации с ДНК на РНК. Делеция происходит во время мейоза при сперматогенезе или овогенезе.

Вилочковая железа располагается в переднем средостении и отвечает за Т-клеточное звено иммунитета. Тимусный эпителий у больных не обеспечивает нормальное развитие Т-клеток, в результате чего страдает клеточный и гуморальный иммунитет. При гипоплазии органа возникает первичный иммунодефицит и образуются неполноценные Т-лимфоциты. Эти клетки крови вырабатываются лейкоцитарным ростком красного костного мозга и мигрируют в тимус. В норме они распознают чужеродные белки и устраняют их.
К факторам риска, провоцирующим развитие синдрома, относятся патологии беременной женщины:
- сахарный диабет,
- употребление спиртных напитков,
- вирусные инфекции в первом триместре,
- ЧМТ,
- прием запрещенных фармпрепаратов,
- воздействие химических веществ.
Патогенетические звенья синдрома:
- микроделеция специфических последовательностей ДНК в области 22 хромосомы,
- мутация генов,
- нарушение дифференцировки стволовых клеток,
- нарушение формирования 3 и 4 глоточных карманов или фарингеальных мешков,
- дисфункция околощитовидных желез,
- пороки сердца,
- лицевые мальформации.
Синдром Ди Джорджи – изолированный Т-клеточный дефицит без определения клеточного иммунитета. При этом специфические антитела вырабатываются на очень низком уровне. У больных вирусные инфекции протекают в тяжелой форме. Пороки развития лицевых структур сочетаются с широким спектром врожденных пороков сердца и поражением дуги аорты.
Симптоматика
Первые симптомы патологии появляются сразу после рождения ребенка. Восстановление Т-клеточного иммунитета наблюдается у детей, переживших 6-месячный возраст.

Клинические признаки синдрома:
- Невооруженным глазом можно обнаружить аномалии лица, к которым относятся: расщепление неба, “готическое» небо, микрогнатия верхнечелюстных костей, «рыбий» рот, маленький нос с широкой переносицей, деформированные и низко расположенные ушные раковины, микроцефалия, широко расставленные глаза, косоглазие, наличие эпиканта, специфический разрез глаз с опущением наружных уголков.
- Проявления врожденных пороков сердца также выступают на первый план. У больных появляются признаки сердечной недостаточности: акроцианоз, тахикардия, одышка после физической нагрузки. Подобные процессы в организме больного ребенка требуют оказания квалифицированной медицинской помощи. В противном случае может развиться сердечная недостаточность или наступить ранняя смерть.
- Гипоплазия паращитовидных желез приводит к гипокальциемии и появлению у детей судорог и тетании, которые возникают при снижении концентрации в крови паратгормона. Судорожный синдром гипокальциемического типа возникает в первые дни после рождения ребенка, не купируется противосудорожными препаратами и нередко приводит к смерти младенца.
- Первичный иммунодефицит – следствие гипоплазии вилочковой железы. Ослабление естественной резистентности организма приводит к затяжным и тяжелым инфекционным заболеваниям, плохо поддающимся стандартной противомикробной терапии. У больных детей обнаруживается явная тенденция к инфекциям верхних дыхательных путей, протекающим в виде ринофарингита, отита, бронхопневмонии; дигестивным и кожным инфекциям, протекающим в виде диареи и пиодермии соответственно.
- При поражении ЦНС частично атрофируется кора головного мозга, возникает гипоплазия мозжечка, что проявляется нарушением походки, парезами и параличами, изменением чувствительности. Возможно у больных развитие умственной отсталости и возникновение неврологических расстройств, которые проявляются у детей с первых дней жизни. У ребят постарше отмечается беспокойство и психоэмоциональная лабильность. Психиатрическая патология у подростков проявляется синдромом гиперактивности, шизофренией, маниакально-депрессивным психозом.
- Аномалии пищеварительной системы – укороченный пищевод, отсутствие ануса; дыхательной системы – укорочение и сужение гортани, глотки, трахеи.
- Патология глаз проявляется изменением передней камеры глаза, колобомой, аномалией сосудистой оболочки и сетчатки.
- Со стороны мочевыделительной системы возникают следующие изменения: гидронефроз, атрофия почек, рефлюкс мочи в почечные чашечки и лоханки.
- Поражение костной системы включает аномалии скелета и зубов. Больные рождаются с полидактилией и отсутствием ногтей. У них поздно прорезываются зубы, возникают спонтанные переломы костей, нарушается правильное развитие зубной эмали, развивается кариес.
- Среди прочих проявлений синдрома выделяют: ларингомаляцию, трахеомаляцию, гастроэзофагальный рефлюкс, глухоту, нарушение глотания, паховые грыжи.

Разнообразные клинические проявления патологии могут возникать у больных одновременно, сочетаться друг с другом или даже отсутствовать. Нередко синдром проявляется лишь недостаточностью иммунных механизмов.
Оппортунистические инфекции редко угрожают жизни детей с синдромом Ди Джорджи. Обычно у них возникают рецидивирующие отиты и синуситы, обусловленные не только снижением иммунной защиты, но и аномальным строением лицевого скелета.
У пациентов с синдромом Ди Джорджи в крови появляются Т-клетки, обладающие аутоагрессией, что проявляется развитием аутоиммунных заболеваний – цитопении, аутоиммунного тиреоидита, ювенильного ревматоидного артрита, аутоиммунной гемолитической анемии. У них повышен риск образования онкопатологий.
Диагностика
Диагноз синдрома Ди Джорджи ставят после выслушивания жалоб больного, сбора анамнеза жизни и болезни, проведения ряда диагностических процедур.
- В анамнезе больных — частые и тяжелые инфекционные заболевания, разрушение зубов, переломы костей, дисфункция сердца, нарушение психомоторного развития, косоглазие.
- Во время визуального осмотра врач определяет характерные изменения лицевого скелета и черепно-лицевые дисморфии, при аускультации слышит специфические шумы в сердце.
- Иммунограмма – снижение Т- лимфоцитов и иммуноглобулинов, диссоциация между снижением Т-клеток и повышением В-лимфоцитов.
- В крови — лимфопения, гипокальциемия, гиперфосфатемия, гипогаммаглобулинемия.
- УЗИ, МРТ и рентген органов средостения подтверждает отсутствие тимуса.
- Эхокардиография — аномалии сердечно-сосудистой системы.
- Генетическое исследование – метод гибридизации ДНК или мультиплексной ПЦР.
- Амниоцентез – инвазивная процедура, позволяющая выявить микроделеционный синдром до рождения ребенка. Этот метод пренатальной диагностики считается очень травматичным и может закончиться преждевременными родами. ДНК-тест имеет точность 99%. Из крови беременной женщины выделяют ДНК плода и изучают на наличие хромосомных аномалий. Пренатальная диагностика позволяет выявить генетические отклонения у плода и решить вопрос относительно исхода беременности.
На основании полученных данных диагноз патологии можно поставить младенцу уже в родильном доме. Комплексная диагностика позволяет определить тяжесть заболевания, прогнозировать дальнейшую жизнь пациента и назначить грамотное лечение.
Терапевтические мероприятия
Синдром Ди Джорджи — хромосомная аномалия, вылечить которую полностью невозможно. С помощью паллиативных и симптоматических методик специалисты стараются улучшить качество жизни больных и не допустить развития тяжелых осложнений.
Медикаментозная терапия:
- Антибиотики широкого спектра действия — макролиды «Азитромицин», фторхинолоны «Ципрофлоксацин», цефалоспорины «Цефотаксим», защищенные пенициллины «Амоксиклав».
- Противовирусные препараты — «Цитовир», «Ремантадин», «Ацикловир».
- Антимикотические средства — «Кетоконазол», «Флюконазол», «Нистатин».
- Иммунозаместительная терапия — внутривенное введение донорских иммуноглобулинов.
- Препараты кальция – «Кальций Д3 никомед», «Кальцемин».
Хирургическое лечение заключается в устранении врожденных пороков сердца и трансплантации больным детям вилочковой железы. Пересадка тимуса эффективна только после кардиохирургического вмешательства.
Виды операций по пересадке фетального тимуса:
- орган фиксируют в мышце передней брюшной стенки,
- взвесь эндокринных клеток вводят внутривенно или внутрибрюшинно,
- небольшой фрагмент органа вводят внутрибрюшинно.
Трансплантация плодной ткани тимуса и паращитовидных желез — единственно эффективный метод лечения патологии. Для коррекции пороков развития лица и неба проводят пластические операции.
Правильная педагогическая и психологическая помощь больным детям позволяет нивелировать задержку интеллектуального развития.
Профилактика
Рекомендации специалистов, позволяющие предупредить развития тяжелых осложнений неизлечимого синдрома:
- избегать стрессов, не конфликтовать, иметь оптимистический настрой;
- не переохлаждаться, одеваться по сезону;
- не контактировать с инфекционными больными;
- беременным женщинам не употреблять алкоголь и не курить;
- своевременная вакцинация от вирусных инфекций позволяет сохранить жизнь и здоровье беременным женщинам и плоду.
Прогноз при синдроме Ди Джорджи неоднозначный. Он определяется выраженностью и степенью коррекции дисфункций сердца и эндокринных желез. В большинстве случаев он является крайне неблагоприятным. При отсутствии адекватного лечения дети погибают на первом году жизни от патологии сердца и тяжелых инфекций. Регулярное применение лекарств и особый образ жизни больных увеличивают продолжительность жизни. Но несмотря ни на что, больные дети редко доживают до 10-летнего возраста.
Синдром Ди Джорджи – это генетически детерминированный иммунодефицит, сопровождающийся аномальным строением лица и врожденными пороками сердца. Описанная патология диагностируется сразу после родов, имеет неблагоприятное течение и часто заканчивается смертью младенца. Специалисты рекомендуют будущим родителям взвесить свои моральные и материальные ресурсы прежде, чем родить больного ребенка.
Видео: лекция по синдрому Ди Джорджи
Видео: ребенок с синдромом Ди Джорджи
Источник