Синдром дениса драша что это

Синдром Дениса-Драша является редким состоянием, которое поражает почки и гениталии. Синдром Дениса-Драша характеризуется развитием заболеваний почек, которые развиваются в течение первых нескольких месяцев жизни. Пострадавшие лица развивают такое состояние, которое называется диффузным гломерулосклерозом, при котором рубцовая ткань начинает распространяться по всем клубочкам почек, которые в свою очередь являются крошечными кровеносными сосудами, фильтрующими отходы из крови. У людей с синдромом Дениса-Драша часто развивается почечная недостаточность уже в детском возрасте. Люди с синдромом Дениса-Драша также имеют 90-процентный шанс развития редкой формы рака почки, известной как опухоль Вильмса. Более того, пострадавшие лица могут развивать множественные опухоли в одной или в обеих почках.
Хотя лица мужского пола с синдромом Дениса-Драша имеют типичный набор мужских хромосом (46, XY), у них часто встречаются дисгенезии гонад, при которой наружные половые органы выглядят немного не так как должны или они выглядят явно женскими или вполне женскими. Яички часто не выходят в нужное место. В результате, лица мужского пола с синдромом Дениса-Драша, как правило, не в состоянии иметь биологических детей (бесплодность). А вот в отличие от пострадавших мужского пола, лица женского пола обычно имеют нормальные половые органы и имеют только проблемы с почками.
Синдром Дениса-Драша. Причины
Мутации в гене WT1 вызывают развитие синдрома Дениса-Драша. Ген WT1 кодирует белок, который регулирует активность других генов путем присоединения (связывания) с конкретными участками ДНК. На основе этого действия, белок WT1 называется фактором транскрипции. Белок WT1 играет важную роль в развитии почек и половых желез (яичников у женщин и яичек у мужчин) до рождения.
WT1 генные мутации, которые вызывают синдром Дениса-Драша, приводят к производству ненормального белка, который не может связываться с ДНК. В результате, активность определенных генов является нерегулируемой, что только будет ухудшать развитие почек и половых органов. Аномальное развитие этих органов приведет к развитию диффузного гломерулосклероза и дисгенезии гонад, что очень характерно для синдрома Дениса-Драша. Также, аномальная активность гена, которая вызывает потерю нормальной функции белка WT1, увеличивает риск развития опухоли Вильмса.

Почка пациента с синдромом Дениса-Драша. Опухоль Вильмса выталкивает нормальную почечную паренхиму в сторону.
Хорошо разграниченная опухоль.

Опухоль Вильмса у пациента с синдромом Дениса-Драша.
Подробнее о мутациях и их роли.
Более 96% людей с клиническим диагнозом синдрома Дениса-Драша имеют мутацию в гене WT1. По крайней мере, сегодня уже известно о 80 типах мутаций в гене WT1, все они могут вызвать синдром Дениса-Драша. Подавляющее большинство этих мутаций являются миссенс мутациями в экзонах 9 или 8, которые кодируют очень важные соединения называемые цинковыми пальцами.
- Аминокислота которая кодируется экзоном 9, аргинин-394, имеет решающее значение в определении активности связывания ДНК с белками WT1, мутация именно в экзоне 9 считается самой часто встречающейся у лиц с синдромом Дениса-Драша. Мутация аргинина-394 присутствовала у 14 из 30 пациентов с синдромом Дениса-Драша в одном из исследований.
- Увеличение числа мутаций в других экзонах также было описано, также это касается и типов мутаций. Например, пациенты с делециями имеют высокий шанс развития двусторонней опухоли Вильмса по сравнению с пациентами с миссенс мутациями (52% против 17% соответственно).
- Мутантный цитоплазматический белок WT1, как полагают, может поглощать некоторые WT1 белки дикого типа в цитоплазме, что приводит к уменьшению количества ядерного белка WT1.
- Мутации только в одном из двух генов WT1 будет достаточно для развития нефропатии и интерсексуальных расстройств. Одна из гипотез предполагает то, что ненормальный белок WT1 может взаимодействовать с непораженным диким белком типа WT1. Это событие имеет доминантно-негативный признак.
- Подавляющее большинство пациентов с синдромом Дениса-Драша — лица мужского пола и у них часто присутствует широкий спектр половых аномалий и наоборот, люди с синдромом Денис-Драша женского пола имеют менее серьезные половые аномалии или они у них вообще отсутствуют.
- Развитие опухоли Вильмса у пациентов с болезнью Дениса-Драша является результатом мутаций в двух генах WT1. Опухоль Вильмса является следствием двух независимых событий, которые приводят к потере функций обоих генов WT1. Согласно одной теории, конституционные или зародышевые мутации в одном гене WT1 могут привести к развитию нефротомии (как было описано выше), а вот если произойдет соматическая мутация во втором гене, то это вызовет неконтролируемую клеточную пролиферацию и образование опухоли Вильмса.
Синдром Дениса-Драша. Патофизиология
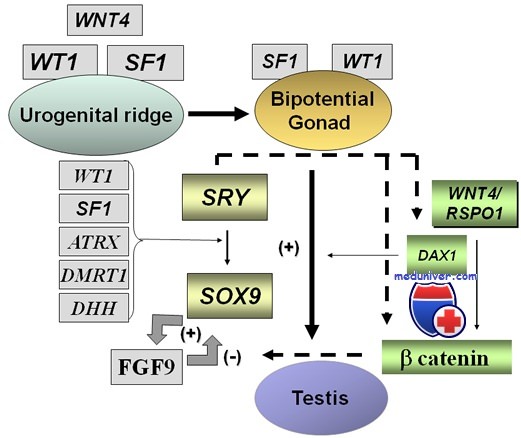
Синдром Дениса-Драша, как мы уже говорили, является результатом мутаций в гене WT1, который расположен на полосе хромосомы 11p13. Белок WT1 (который кодируется одноименным геном) является фактором транскрипции, который преимущественно экспрессируется в эмбриональных клетках почек и в половых железах. Ген WT1 содержит состоит из 10 экзонов, экзоны 1-6 кодируют регулирующий домен, который регулирует экспрессию генов-мишеней, а экзоны 7-10 кодируют четыре цинковых пальца ДНК-связывающей области белка WT1. Белок WT1 опосредует мезенхимально-эпителиальный переход и дифференцировку во время морфогенеза почек и половых желез, подавляя гены, кодирующие факторы клеточной пролиферации и активации генов. И именно точечные мутации в гене WT1 (особенно в экзонах 5,8,9) могут привести к потере его регулирующей функции, что приведет к развитию последующих отклонений в клубочковом формировании и дифференциации половых желез, которые часто встречаются при синдроме Дениса-Драша. Другие, более тяжелые хромосомные аномалии, включают полные делеции группы 11p13, в результате такой аберрации у человека развиваются опухоли Вильмса, аниридии, мочеполовые аномалии и умственная отсталость.
Синдром Дениса-Драша. Симптомы и проявления
У женщин, внутри которых развивается ребенок с синдромом Дениса-Драша, особенно с уклоном на нефропатию, плацента, беременность и роды будут, как правило, нормальными. В начале (обычно в течение первого года жизни), типичные проявления будут включать нефротический синдром (например, отек, вздутие живота, рецидивирующие инфекции). С быстрым снижением скорости клубочковой фильтрации и прогрессированием хронической почечной недостаточности, у человека будут развиваться следующие дополнительные симптомы:
- Снижение активности
- Плохой аппетит и рост
- Потеря этапов развития
- Неспецифические боли
- Олигурия
Знаки синдрома Дениса-Драша, связанные с опухолью Вильмса включают увеличение брюшной полости, это является наиболее распространенным проявлением, которое встречается у 90% детей при обычном осмотре. Другие симптомы и проявления включают в себя:
- Боль в животе
- Гематурия
- Потеря веса
- Плохой аппетит
- Паховая грыжа
Физическое обследование
Признаки, связанные с нефропатией включают знаки развития нефротического синдрома (например, отеки, асцит). Свидетельства развития венозного тромбоза иногда могут развиться вторично к нефротическому синдрому. Бледность может отражать наличие анемии. Если присутствует хроническая почечная недостаточность, то следующие признаки могут развиваться вторично по отношению к почечной остеодистрофии:
- Снижение активности
- Замедление развития
- Снижение темпов роста
- Скелетные аномалии (аналогичные тем, которые встречаются при дефиците витамина Д)
Признаки, связанные с опухолью Вильмса включают ощутимую массу в брюшной полости, гематурию, и гипертонию. Другие признаки могут включать в себя:
- Потеря веса
- Обстипация
- Паховая грыжа
- Дыхательная недостаточность от плеврального выпота
- Признаки застойной сердечной недостаточности
Признаки, связанные с интерсексуальными нарушениями
- Большинство мужчин с синдромом Дениса-Драша с кариотипом 46, XY или 46, XX / 46, XY развивают псевдогермафродитизм. Спектр аномалий наружных половых органов может включать гипоспадии с крипторхизмом, увеличенный клитор, слияние губ, раздвоенность мошонки с пальпированием половых желез и микропенис.
- Аномалии внутренних половых органов у этих лиц также могут включать наличие влагалища и матки.
Синдром Дениса-Драша. Диагностика
Общий анализ мочи – протеинурия является признаком нефропатии, которая развивается при синдроме Дениса-Драша. Гематурия также может быть выявлена.
- Почечные функциональные тесты: уровень АМК и уровень креатинина могут быть в пределах референсных значений на ранних стадиях синдрома Дениса-Драша. Эти уровни могут ухудшиться с наступающей нефропатией или двусторонним развитием опухоли Вильмса. Терминальная стадия заболевания почек сопровождается гиперкалиемией и гиперфосфатемией.
- Опухолевые маркеры Вильмса: повышенные уровни гиалуроновой кислоты, повышенная активность гиалуроновой кислоты, эритропоэтина, и ренин прогормона связаны с опухолью Вильмса.
- Хромосомный анализ может понадобиться для определения типа мутации.
Визуализация
Брюшное и тазовое УЗИ. Его стоит выполнять у всех пациентов, которые жалуются на симптомы предлагающие наличие синдрома Дениса-Драша. Через равные промежутки времени после первоначального диагноза, врачам необходимо продолжать проводить УЗИ почек для выявления опухоли Вильмса. УЗИ – неплохой метод, но его сегодня все чаще заменяют на КТ или МРТ.
Синдром Дениса-Драша. Лечение
Краеугольным камнем в лечении синдрома Дениса-Драша является медикаментозная терапия, которая должна включать в себя контроль баланса жидкости и электролитов, лечение гипертонии, почечные терапии для пациентов с терминальной стадией почечной недостаточности или для пациентов после двусторонней нефрэктомии и химиотерапии.
Хирургическое лечение больных с синдромом Дениса-Драша и опухолью Вильмса включает следующие аспекты:
- Для пациентов без опухоли Вильмса, в момент первой диагностики синдрома Дениса-Драша, будет необходима ранняя профилактическая двусторонняя нефрэктомия с последующей заместительной почечной терапией, это необходимо для того, чтобы можно было избежать повышения риска развития опухоли Вильмса, и это, как следствие, может стать той причиной по которой пострадавшему не проведут трансплантацию почки. Так как риск риск развития злокачественности гонад также высок у пациентов с дисгенезией гонад, хирург может принять решение по выполнению кастрации в момент нефрэктомии. Назначение соответствующей гормональной терапии у этих пациентов в период полового созревания может быть оправданным.
- Если у пациента, на момент диагностики, также уже была обнаружена опухоль Вильмса, то врач может принять решение по проведению трансплантации почки после двусторонней нефрэктомии. Пациенты с опухолью Вильмса еще до операции должны пройти химиотерапию.
Синдром Дениса-Драша. Осложнения
Пациенты с нефротическим синдромом могут столкнуться с развитием рецидивирующих инфекций, недостатком питательных веществ, и, иногда, с венозным тромбозом. Прогрессия к терминальной стадии почечной болезни является неизбежной. Осложнения химиотерапии могут включать развитие угрожающих жизни инфекций и вторичных опухолей. Риск развития опухоли Вильмса в любой остаточной почечной ткани после операции является высоким.
Синдром Дениса-Драша. Прогноз
Все пациенты развивают нефропатию и терминальную почечную болезнь в течение 2 лет. Практически у всех больных с синдромом Дениса-Драша развиваются опухоли Вильмса. Пациенты с односторонней опухолью Вильмса находятся в опасности от развития контралатеральной опухоли.
Источник
Синдром Дени-Дрэша — клиника, диагностикаЭти редкие заболевания вызваны мутациями гена — супрессора опухоли Вильмса (WT1), который кодирует фактор транскрипции, в норме подавляющий образование нефробластомы (опухоли Вильмса). Проявления синдрома Дени—Дрэша и синдрома Фрайзера во многом схожи (нарастающий диффузный мезангиальный склероз и дисгенезия гонад с кариотипом XY), но поскольку в их основе лежат разные мутации, то для синдрома Фрайзера нехарактерно развитие нефробластомы. Диффузный мезангиальный склероз характерен для синдрома Дени—Дрэша, но может быть и самостоятельным заболеванием. У некоторых больных с изолированным диффузным мезангиальным склерозом также есть мутации гена WT1. Фактор транскрипции WT1 может как подавлять, так и активировать транскрипцию генов, в зависимости от того, с каким участком ДНК он связывается. В свою очередь, другие белки могут изменять его активность. Связываясь с мРНК, WT1 может регулировать экспрессию генов, и на уровне транскрипции, и на посттранскрипционном уровне. В период формирования почки WT1 экспрессируется в конденсирующейся мезенхиме, в образующихся при конденсации мезенхимы везикулах и подоцитах, в окончательной почке его экспрессия сохраняется в подоцитах. Такой характер экспрессии наводит на мысль, что WT1 участвует в дифференцировке клеток мезенхимы в подоциты и поддерживает их нормальный фенотип. Нарушение функции WT1 ведет к неконтролируемому росту метанефрогенной ткани и образованию нефробластомы или нарушению дифференцировки подоцитов. Фактор транскрипции WT1 экспрессируется также в клетках полового тяжа, гранулезных клетках яичниковых фолликулов и клетках Сертоли. Возможно, нормальная дифференцировка половых желез также зависит от фактора транскрипции WT1. Для синдрома Дени—Дрэша характерны: нефробластома, дисгенезия гонад с кариотипом XY и наружными половыми органами промежуточного типа и гломерулопатия. Последняя обычно проявляется нефротическим синдромом, возникающим в первый год жизни и приводящим к развитию ХПН к 3 годам. У детей синдром Дени—Дрэша может быть как полным, так и неполным (гломерулопатия и аномалии половых органов без нефробластомы или гломерулопатия и нефробластома без аномалий половых органов). Как и при других формах дисгенезии гонад с кариотипом XY, у этих больных повышен риск гонадобластом. Если диагноз синдрома Дени—Дрэша не вызывает сомнений, то, учитывая высокий риск нефробластомы, показана двусторонняя нефрэктомия. Для больных с синдромом Дени—Дрэша характерно специфическое поражение почек — диффузный мезангиальный склероз. Поражаются интракортикальные нефроны, в них расширяется мезангий и облитерируются капилляры клубочка, из-за чего клубочек превращается в бесформенную массу матрикса с венцом из гипертрофированных подоцитов. Причина заболевания — миссенс-мутация в гене WT1, приводящая к замене аргинина на триптофан в положении 394. Реже встречается замена аспартата на аспарагин в положении 396 и аргинина на гистидин в положении 366. В результате этих мутаций фактор транскрипции WT1 теряет способность связываться с ДНК. В большинстве случаев эти мутации возникают впервые, хотя могут и наследоваться. Хотя больные с синдромом Дени—Дрэша являются носителями лишь одного мутантного рецессивного аллеля гена WT1, но в нефробластоме поражены оба аллеля, причем каждый аллель несет разные мутации. При синдроме Дени—Дрэша в почках остаются очаги метанефрогенной ткани, несущие унаследованную мутацию гена WT1 в одном из аллелей. Если в клетках этой ткани происходит мутация второго аллеля, то это приводит к неконтролируемой пролиферации клеток и возникновению нефробластомы. Механизм развития диффузного мезангиального склероза и дисгенезии гонад при синдроме Дени—Дрэша иной, чем при нефробластоме, поскольку больной гетерозиготен и несет лишь один мутантный аллель гена WT1. Эта мутация проявляется как доминантно-негативная, то есть дефектный белок нарушает функцию продукта нормального аллеля, лишая его способности контролировать экспрессию других генов. Доминантно-негативный характер мутации гена WT1 объясняет, почему поражение почек и аномалии строения наружных половых органов при синдроме Дени—Дрэша более тяжелые, чем при синдроме WAGR, при котором в результате делеции или мутации со сдвигом рамки считывания один аллель гена WT1 становится нулевым, что не мешает экспрессии нормального аллеля.
— Также рекомендуем «Болезнь Фабри — клиника, диагностика» Оглавление темы «Наследственные болезни почек»:
|
Источник
Русский Медицинский Сервер / Лечение в Италии / Центр лечения редких заболеваний в Милане / Синдром Дениса-Драша — лечение в Италии
Синдром Дениса-Драша является редким состоянием, которое поражает почки и гениталии. Синдром Дениса-Драша характеризуется развитием заболеваний почек, которые развиваются в течение первых нескольких месяцев жизни. Пострадавшие лица развивают такое состояние, которое называется диффузным гломерулосклерозом, при котором рубцовая ткань начинает распространяться по всем клубочкам почек, которые в свою очередь являются крошечными кровеносными сосудами, фильтрующими отходы из крови.
У людей с синдромом Дениса-Драша часто развивается почечная недостаточность уже в детском возрасте. Люди с синдромом Дениса-Драша также имеют 90-процентный шанс развития редкой формы рака почки, известной как опухоль Вильмса. Более того, пострадавшие лица могут развивать множественные опухоли в одной или в обеих почках.

Хотя лица мужского пола с синдромом Дениса-Драша имеют типичный набор мужских хромосом (46, XY), у них часто встречаются дисгенезии гонад, при которой наружные половые органы выглядят немного не так как должны или они выглядят явно женскими или вполне женскими. Яички часто не выходят в нужное место. В результате, лица мужского пола с синдромом Дениса-Драша, как правило, не в состоянии иметь биологических детей (бесплодность). А вот в отличие от пострадавших мужского пола, лица женского пола обычно имеют нормальные половые органы и имеют только проблемы с почками.
Мутации в гене WT1 вызывают развитие синдрома Дениса-Драша. Ген WT1 кодирует белок, который регулирует активность других генов путем присоединения (связывания) с конкретными участками ДНК. На основе этого действия, белок WT1 называется фактором транскрипции. Белок WT1 играет важную роль в развитии почек и половых желез (яичников у женщин и яичек у мужчин) до рождения.
WT1 генные мутации, которые вызывают синдром Дениса-Драша, приводят к производству ненормального белка, который не может связываться с ДНК. В результате, активность определенных генов является нерегулируемой, что только будет ухудшать развитие почек и половых органов. Аномальное развитие этих органов приведет к развитию диффузного гломерулосклероза и дисгенезии гонад, что очень характерно для синдрома Дениса-Драша. Также, аномальная активность гена, которая вызывает потерю нормальной функции белка WT1, увеличивает риск развития опухоли Вильмса.
У женщин, внутри которых развивается ребенок с синдромом Дениса-Драша, особенно с уклоном на нефропатию, плацента, беременность и роды будут, как правило, нормальными. В начале (обычно в течение первого года жизни), типичные проявления будут включать нефротический синдром (например, отек, вздутие живота, рецидивирующие инфекции). С быстрым снижением скорости клубочковой фильтрации и прогрессированием хронической почечной недостаточности, у человека будут развиваться следующие дополнительные симптомы:
- Снижение активности
- Плохой аппетит и рост
- Потеря этапов развития
- Неспецифические боли
- Олигурия
Знаки синдрома Дениса-Драша, связанные с опухолью Вильмса включают увеличение брюшной полости, это является наиболее распространенным проявлением, которое встречается у 90% детей при обычном осмотре. Другие симптомы и проявления включают в себя:
- Боль в животе
- Гематурия
- Потеря веса
- Плохой аппетит
- Паховая грыжа
- Физическое обследование
Признаки, связанные с нефропатией включают знаки развития нефротического синдрома (например, отеки, асцит). Свидетельства развития венозного тромбоза иногда могут развиться вторично к нефротическому синдрому. Бледность может отражать наличие анемии. Если присутствует хроническая почечная недостаточность, то следующие признаки могут развиваться вторично по отношению к почечной остеодистрофии:
- Снижение активности
- Замедление развития
- Снижение темпов роста
- Скелетные аномалии (аналогичные тем, которые встречаются при дефиците витамина Д)
Признаки, связанные с опухолью Вильмса включают ощутимую массу в брюшной полости, гематурию, и гипертонию.
! Несмотря на то, что многие из описанных в данном разделе болезней считаются неизлечимыми, в Центре лечения редких заболеваний в Милане постоянно ведется поиск новых методов. Благодаря генной терапии удалось добиться выдающихся результатов и полностью излечить некоторые редкие синдромы.
Обратитесь к консультанту на сайте или оставьте заявку — так вы можете узнать, какие методы предлагают итальянские врачи. Возможно, данное заболевание уже научились лечить в Милане.
+7 (925) 50 254 50 –
срочное лечение в Италии
ЗАПРОС в КЛИНИКУ
Источник