Синдром делеции длинного плеча 22 хромосомы

Другие названия синдрома делеции 22q11.2
- — 22q11DS
- — Аутосомно-доминантный G/BBB синдром Опитца(ADOS)
- — CATCH 22
- — CATCH22
- — Синдром Кайлера
- — Хромосомный синдром делеции 22q11.2
- — Синдром конотрункальных и лицевых аномалий
- — Синдром Ди Джорджи
- — Последовательность Ди Джорджи
- — Микроделеция 22q11.2
- — Моносомия 22q11
- — Синдром Седлакова
- — Синдром Шпринтцена
- — Синдром Такао
- — VCFS
- — Вело-кардио-фациальный синдром
- — Х-сцепленный BBB/G синдром Опитца
- — Х-сцепленный G/BBB синдром Опитца
- — Х-сцепленный синдром Опитца
- — XLOS
Синдром делеции 22q11.2 связан с целым рядом проблем, в том числе: врожденные пороки сердца, аномалии неба, дисфункции иммунной системы, включая развитие аутоиммунных заболеваний, низкое содержание кальция (гипокальциемия) и другие эндокринные патологии, такие, как проблемы с щитовидной железой и дефицит гормона роста, желудочно-кишечные проблемы, трудности в кормлении, аномалии почек, потеря слуха, судороги, скелетные аномалии, незначительные аномалии лица и поведенческие различия. Симптомы и проявления этого состояния чрезвычайно разнообразны, даже среди членов одной семьи.
Синдром делеции 22q11.2. Эпидемиология
Частота развития синдрома делеции 22q11.2 составляет примерно от 1/4000 до 1/6000 человек, но эта частота может быть вполне выше, из-за сильной изменчивости проявлений этого синдрома и из-за того, что многие матери по всему миру или отказываются от скрининга новорожденного или в больницах просто не имеется необходимого оборудования для его проведения.
Синдром делеции 22q11.2. Похожие расстройства
Синдром Смита-Лемли-Опитца. Этот синдром является переменным генетическим расстройством, которое характеризуется медленным ростом до и после рождения, маленькой головой (микроцефалия), умственной отсталостью и множественными врожденными дефектами, включая расщелину нёба, пороки сердца, дополнительные пальцы рук и ног, слаборазвитые наружные половые органы у мужчин.
Синдром Алажилля – это генетическое заболевание печени, которое, как правило, присутствует при рождении. Оно характеризуется недостаточным прохождением желчи из-за более маленького, чем обычно, количества желчных протоков внутри печени. В некоторых случаях, ребенок может родиться вообще без каких-либо желчных протоков. Основные симптомы и проявления включают желтуху, аномалии глаз и сердца, аномальные формы позвонка, сжатие нервов внутри нижней части позвоночника, отсутствие глубоких сухожильных рефлексов, умственную отсталость, аномалии почек и недостаточность поджелудочной железы.
VACTERL ассоциация – неслучайная ассоциация врожденных дефектов, которые затрагивают несколько систем органов. Термин VACTERL – это акроним, каждая буква которого представляет первую букву одной из наиболее распространенных проблем при этом синдроме: (V) = аномалии позвонков, (A) = анальная атрезия, (С) = сердечные дефекты, (Т) = трахеопищеводный свищ, (Е) = атрезия пищевода, (R) = аномалии почек и (L) = аномалии конечностей. Кроме того, пострадавшие дети могут также проявлять недостатки роста и неспособность набирать вес.
Синдром делеции 22q11.2. Причины
У людей с синдром делеции 22q11.2 не хватает небольшой части хромосомы 22. Эта делеция происходит спонтанно у 93% пострадавших и наследуется от родителей у 7% пациентов. Риск передачи аномальной хромосомы от родителей к потомству составляет 50% для каждой беременности. Риск развития этого синдрома одинаков как для мужчин так и для женщин.
Синдром делеции 22q11.2. Симптомы и проявления
.")
Легкий дисморфизм лица у девочек с синдромом делеции 22q11.2 (слева в возрасте 25 лет, справа в возрасте 11 лет).
Нарушения с более чем одним идентификационным признаком называются синдромами. Некоторые из особенностей этого синдрома включают аномалии нёба, аномалии сердца, проблемы с обучаемостью и другие различные физические особенности, о них мы поговорим ниже.
Лица с синдром делеции 22q11.2 обычно имеют «заячью губу» и «волчью пасть». Аномалии сердца, связанные с этим синдромом, могут включать в себя: дефекты в межжелудочковой перегородке, аномалии аорты, аномалии, при которых, блокируется отток крови из правого желудочка сердца в легкие, в результате чего у ребенка будет увеличен правый желудочек и смещена аорта.
Мягкие задержки в обучении присутствуют у большинства людей с синдром делеции 22q11.2. Среднее значение IQ у детей поздних школьных классов составляет 69-87. Проблемы с абстракцией при чтении книг и достаточно значительные проблемы с математикой, как правило, будут очевидны уже в школьном возрасте. А вот задержка умственного развития встречаетcя реже, но она также может присутствовать у некоторых лиц с этим синдромом.
Около четверти пациентов развивают психотические симптомы и большинству из них ставится диагноз шизофрении еще в подростковом возрасте. Клинические исследования показали, что люди с синдром делеции 22q11.2 в 25 раз чаще развивают шизофренические симптомы, чем здоровые люди. Шизофрения, которая связанна с синдром делеции 22q11.2, имеет хронический курс течения. Эта шизофрения также связана с плохим ответом на психиатрические препараты.
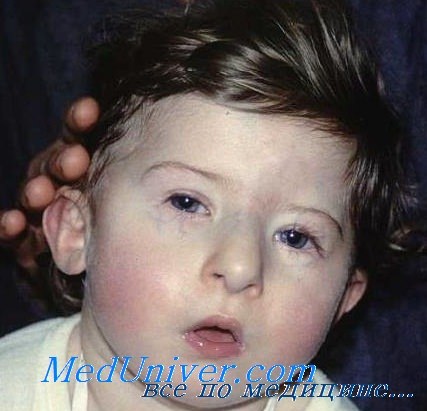
Мальчик с синдромом Ди Джорджи
Среди физических особенностей этого синдрома можно выделить потерю мышечного тонуса (гипотония), небольшой рост, конические руки и пальцы, небольшую окружность головы (микроцефалия), ретрогнатию, трубчатый нос, плоские щеки, длинную верхнюю челюсть, губной желобок, синие пятна под глазами, маленькие уши, толстые внешние диски уха, уши разных размеров и другие проявления.
У некоторых пациентов также могут развиться и другие проявления этого синдрома, но они встречаются относительно редко, к ним можно отнести: недостаточно развитый или отсутствующий тимус, потеря слуха, помутнение хрусталика глаза, катаракта, микрофтальмии, искривление позвоночника (сколиоз), паховые или пупочные грыжи, крипторхизм, дефицит кальция в крови (гипокальциемия), небольшие аденоиды или небольшие миндалины. Кроме того, новорожденные дети могут иметь некоторые проблемы с дыханием из-за утопленной челюсти и из-за плохого мышечного тонуса в области горла.
Синдром делеции 22q11.2. Диагностика
Синдром делеции 22q11.2 будет подозреваться только при наличии характерных клинических симптомов и проявлений. Подтверждение диагноза осуществляется хромосомным анализом (наличие делеции в хромосоме 22). На сегодняшний день существует большое количество новых тестов, которыми можно выявить эту делецию, в том числе, SNP, геномная гибридизация и MLPA , FISH. Благодаря этим методам, сегодня можно подтверждать наличие делеции в 22-й хромосоме у абсолютного большинства пациентов.
Синдром делеции 22q11.2. Лечение
Командный подход часто оказывается полезным и в целом, он рекомендуется при принятии решений о лечении симптомов и проявлений у пациентов с синдромом делеции 22q11.2. Педиатр, логопед, ортодонт, пластический хирург, отоларинголог, сурдолог, стоматолог, кардиолог, иммунолог, эндокринолог, гастроэнтеролог, уролог / нефролог, ортопед и другие специалисты должны действовать вместе для достижения лучших результатов в лечении. Однако, стоит уяснить один факт – причину развития этого синдрома сегодня еще невозможно вылечить, поэтому все методики будут направленны только на контроль и устранение некоторых аномалий, в первую очередь это касается сердца.
Источник

Широко известно, что гиппокамп играет решающую роль в процессах обучения, воображения и памяти. Результаты различных МРТ исследований и посмертных вскрытий позволяют предполагать, что у пациентов с шизофренией снижен объем гиппокампа. Также обнаружено уменьшение гиппокампа у пациентов с расстройствами шизофренического спектра, включая первый психотический эпизод, а также у участников с высоким и супервысоким риском развития психоза. Однако недавний мета-анализ показал, что не было никаких доказательств значительного уменьшения объема всего гиппокампа у пациентов с клиническим высоким риском развития психоза. В связи с этим было высказано предположение о том, что на ранних стадиях заболевания могут присутствовать более тонкие изменения в гиппокампе. Такие нарушения обнаружены у лиц со сверхвысоким риском развития психоза.
Сегодня существует несколько теорий, объясняющих взаимосвязь между психозом и снижением объема гиппокампа. Особое внимание уделяется функциональной анатомии. Каждое поле гиппокампа имеет разную плотность пирамидных нейронов, разнообразную синаптическую структуру и особые способы связи с кортикальными областями. Примечательно, что зубчатая извилина, СА3 и СА1 являются частью трисинаптического пути и отвечают за кодирование эпизодической памяти, тогда как основание гиппокампа отвечает за постоянство такой информации и ее передачу в неокортекс.
Подписывайтесь на нас на Patreon, и мы будем выпускать ещё больше интересных материалов
На данный момент нет единого мнения насчёт того, как зона занимает центральное место в развитии психоза. Одна теория утверждает, что гиперактивность поля CA3, которое ответственно за кодирование и извлечение памяти, может участвовать в генерации ложных воспоминаний, воспринимаемых как галлюцинации больных шизофренией. С другой стороны, CA1 оказался самым ранним пораженным участком у пациентов с первым эпизодом психоза и сверхвысоким риском его развития.
В целом, снижение объема гиппокампа было показано в поперечных и продольных исследованиях у пациентов с клиническим риском развития психоза. У лиц, имеющих генетический риск (например, братья и сестры пациентов), также были выявлены изменения гиппокампа, что позволяет судить о наследуемом характере аномалии. Ведутся дебаты относительно того, является ли снижение объема гиппокампа причиной или следствием психотических симптомов, а также может ли этот процесс быть более сложным, взаимозависимым комплексом.
В связи с этим изучение населения с генетическим риском развития психоза дает уникальную возможность проводить оценку пациентов с детства, выяснять временные взаимосвязи между развитием гиппокампа и началом психоза.
Синдром делеции 22-й хромосомы (22q11DS) – это расстройство развития нервной системы, которое является следствием делеции длинного плеча 1-й копии 22-й хромосомы. Данный синдром считается наиболее важным генетическим фактором в развитии психоза. Поскольку среди пациентов с 22q11DS у 41% развивается психотическое расстройство в зрелом возрасте, синдром признан ценной моделью для выявления ранних биомаркеров психоза. У носителей делеции наблюдается снижение когнитивных способностей и способности к обучению, они более склонны к развитию психических расстройств, таких как синдром дефицита внимания и гиперактивности (СДВГ), тревожность и обсессивно-компульсивное расстройство (ОКР). Широкий спектр медицинских состояний, включающих врожденный порок сердца, гипоплазии (аплазии) тимуса и гипоплазию паращитовидной железы. Аномалии мозга также являются общей особенностью синдрома, так как у 11% пациентов наблюдается общее уменьшение объема мозга и снижение процесса образования извилин в лобной и теменной доли.
Синдром делеции 22-й хромосомы (22q11DS) оказался отличной моделью для проспективного исследования лиц с высоким риском развития шизофрении, чтобы раскрыть патофизиологические процессы, предшествующие началу психоза. Повторные МРТ мозга были получены от 140 пациентов с 22q11DS (53 были с умеренными и тяжелыми психотическими симптомами) и 135 результатов от участников контрольной группы. Участники исследования были отобраны в Женеве в 2001 году, их возраст варьировался от 6 до 35 лет, обе группы были сопоставимы по полу и возрасту. Анализ гиппокампа проводили с использованием FreeSurfer-v. 6 и FIRST-FSL. Сравнивались объемы всего гиппокампа и его полей у разных групп.
По результатам исследования, при сравнении с контрольной группой у пациентов с 22q11DS объем всех полей был значительно меньше, за исключением полей CA2/3, но дивергентных траекторий в развитии гиппокампа обнаружено не было. Кроме того, исследования показали, что объем гиппокампа неизменно меньше у всех пациентов с 22q11DS, включенных в исследование с возраста 6 лет.
При сравнении пациентов с 22q11DS, у которых обнаруживались психопатологические симптомы, с пациентами с 22q11DS без психоза, обнаружено уменьшение объема гиппокампа в позднем подростковом возрасте, начиная с CA1 и распространяясь на другие поля. Было продемонстрировано, что у пациентов с 22q11DS и психотическими симптомами происходит дальнейшее снижение объема в подростковом возрасте – уязвимом периоде для возникновения психоза. Примечательно, что поля CA2/3 были меньше у пациентов с психотическими симптомами, однако являлись единственной областью, не измененной у пациентов с 22q11DS без психотических симптомов, по сравнению с контрольной группой. Это позволяет предположить, что атрофия этой области значительно коррелирует с наличием позитивных психотических симптомов.
При исследовании пациентов с 22q11DS не было доказано, что степень выраженности психотических симптомов может быть связана со степенью потери объема гиппокампа. Тем не менее были найдены зоны гиппокампа, которые изменяются с позднего подросткового возраста. В связи с этим было высказано предположение о том, что существует связь между прогрессированием заболевания и уменьшением объема гиппокампа. Это несоответствие может быть объяснено тем, что позитивные психотические симптомы имеют по своей природе колеблющийся характер, который не обязательно параллелен общему прогрессированию заболевания, особенно у пациентов с 22q11DS.
Стоит отметить, что в проводимом исследовании объемы большинства областей гиппокампа, такие как его основание, CA1, CA4 и зубчатая извилина, которые, как ожидается, могут играть роль в развитие психоза, уже были снижены во всей группе с 22q11DS по сравнению с контрольной группой. Поэтому нельзя игнорировать тот факт, что наличие 22q11DS само по себе является фактором риска развития психоза.
Таким образом, было продемонстрировано в большой выборке пациентов с 22q11DS уменьшение объема гиппокампа по сравнению с контрольной группой, предполагая, что это может быть анатомической чертой синдрома. Касательно полей гиппокампа, СА1 был первым пораженным участком, в то время как CA3 был последним, и его атрофия была исключительно связана с появлением позитивной симптоматики.
Учитывая тот факт, что больше нет продольных исследований, которые могли бы оценить возникновение позитивных симптомов и изменений объема гиппокампа за такой долгий период времени, а также наличие изменений в гиппокампе у здоровых родственников пациентов с шизофренией, ученые считают, что будущие исследования должны рассматривать вопрос о наличии аномалий или сниженного объема гиппокампа с раннего возраста у лиц, не имеющих психотических проявлений, однако у которых в последствии может развиться шизофрения.
Автор перевода: Сафи А. И.
Источник: Valentina Mancini, Corrado Sandini, Maria C. Padula et al. Positive psychotic symptoms are associated with divergent developmental trajectories of hippocampal volume during late adolescence in patients with 22q11DS [Published: 04 June 2019]
Источник
Синдром Ди Джорджи (вело-кардио-фациальный синдром, синдром делеции 22q11) — этиология, клиника, диагностикаСиндром Ди Джорджи (вело-кардио-фациальный синдром, синдром делеции 22q11) связан с дефектом развития четвертого глоточного кармана и четвертой жаберной дуги. Данный синдром имеет генетически неоднородный характер с частичной моносомией проксимального плеча 22-й хромосомы (при микроскопическом исследовании выявляется в трети случаев) и высокой частотой делеций (88%), выявленных по результатам FISH-исследований (Driscoll et al., 1993). В редких случаях заболевание сочеталось делецией 17р и 10p (Levy-Mozziconacci et al., 1994). Уровень распространения синдрома делеции 22q11 (22Q11DS оценивается приблизительно как 3 случая на 10000 новорожденных (Oskarsdottir 2005) и возможно несколько чаще встречается среди девочек (личные неопубликованные данные). В настоящее время большая часть практикующих врачей рассматривает синдром делеции 22q11 как общепринятый синдром с фенотипом Ди Джорджи. Таким образом, диагноз устанавливается в случае выявления «САТСН-22 фенотипа» (аномалии сердца (Cardiac), аномалии (Anomalous) лица, гипоплазии тимуса (Thymus), расщелины (Cleft) неба (включая только подслизистое расщепление) и гипокальциемии (Hypocalcaemia) и делеции сегмента 22q11, выявленной методом FISH-диагностики. Анализ методом FISH обеих метафазных хромосом и интерфазной клетки пациента с синдромом Ди Джорджи, а) Клинические проявления. Тимус и паратиреоидные железы отсутствуют или эктопированы, также часто отмечается мальформация крупных сосудов основания сердца. Отсутствие тимуса может сопровождаться тяжелым иммунодефицитом, а отсутствие паращитовидных желез—тяжелыми гипокальциемическими судорогами в период новорожденности и позже. Гипокальциемия связана с паратиреоидным гормоном. Часто встречается расщелина неба, микрогнатия, низко расположенные уши и гипертелоризм (Conley et al., 1979). Частично совпадающий фенотип, известный как вело-кардио-фациальный синдром или синдром Шпринтцена, проявляется сходными аномалиями сердца, дисморфизмом лица, расщелиной неба и задержкой умственного развития, иногда сопровождается поздним развитием психиатрических отклонений и также сочетается с делецией 22q11. Также может отмечаться гипоплазия мозжечка (Devriendt et al., 1996). Синдром делеции сегмента 22q11 проявляется достаточно типичным поведенческим фенотипом. Отмечается задержка развития разговорной речи, но некоторые вербальные навыки, в частности словарный запас, имеют тенденцию к лучшему развитию, чем функции, связанные с достижением школьного возраста. Коэффициент IQ обычно составляет около 70, у отдельных пациентов превышает 100 и (очень редко) не достигает 40. Неуклюжесть движений является типичным проявлением данного заболевания, и клинические проявления синдрома делеции сегмента 22q11 часто полностью соответствуют критериям нарушения развития координации.
Характерен упадок сил, который в большинстве случаев отмечается с самого раннего возраста, в связи с чем для начала любых даже самых простых действий пациента необходимо «заводить», что противоречит относительно «хорошему» (по сравнению с другими синдромами с поведенческим фенотипом) IQ. Большая часть пациентов «не успевает» в школе; это касается даже немногочисленной группы с IQ около 100 (Oskarsdottir et al., 2005). У многих пациентов выявляются критерии синдрома гиперактивности с дефицитом внимания (СГДВ), в частности, смешанного подтипа и подтипа с дефицитом внимания. С возрастом у большей части пациентов подтип СДГВ сменяется на подтип с дефицитом внимания (Niklasson et al., 2005). Типичны аутистические проявления (Campbell et al„ 2006), но истинный аутизм встречается редко. Даже в случаях, когда признаки аутизма отсутствуют, у пациентов отмечаются социальные проблемы и проблемы общения. Данные отклонения часто сопровождаются растягиванием слов, проблемами артикуляции и бедной мимикой, характерной для синдрома делеции сегмента 22q11, так как во многих случаях отмечается небно-глоточная недостаточность. Депрессивное настроение часто отмечается, начиная с подросткового возраста. Психозы могут развиваться в позднем подростковом возрасте и раннем взрослом возрасте, даже в случаях с минимальными психиатрическими отклонениями в детском возрасте (Sobin et al., 2005). СДГВ и расстройства группы аутизма при синдроме делеции сегмента 22q11 могут быть результатом патологии головного мозга с уменьшением серого вещества коры, большого хвостатого ядра и аномалии белого вещества лобных долей и мозжечка. В подростковом и взрослом возрасте увеличивается риск развития шизофрении (Niklasson et al., 2002). б) Лечение синдрома Ди Джорджи. Как и в случае других синдромов с поведенческим фенотипом важен ранний диагноз и обучение семьи психологической помощи. СДГВ в некоторых случаях успешно поддается лечению стимуляторами и методами когнитивно-поведенческой терапии в сочетании со специальными программами образования. Ингибиторы захвата серотонина или комбинированного захвата серотонина и норадреналина можно попробовать при длительных периодах депрессивного настроения или дистимии. Психоз (в случае его развития) может с трудом подаваться лечению, а типичные и атипичные нейролептики, несмотря на их эффективность в отношении психотических симптомов, а иногда и слуховые галлюцинации, часто менее эффективны, чем в случае других психозов у молодых людей. — Также рекомендуем «Синдром Дауна (трисомия 21-й хромосомы) — этиология, клиника, диагностика» Редактор: Искандер Милевски. Дата публикации: 4.12.2018 Оглавление темы «Наследственные синдромы в неврологии.»:
|
Источник