Синдром делеции длинного плеча 15 хромосомы

Синдромы Прадера-Вилли и Ангельмана. ХарактеристикаВозможно, наиболее полно изученные примеры роли геномного импринтинга при болезнях человека — синдромы Прадера-Вилли и Ангельмана. Синдром Прадера-Вилли — сравнительно частый дисморфический синдром, характеризующийся ожирением, чрезмерным и беспорядочным аппетитом, небольшими кистями и стопами, низким ростом, гипогонадизмом и умственной отсталостью. Приблизительно в 70% случаев синдрома наблюдают цитогенетическую делецию, затрагивающую проксимальный отдел длинного плеча хромосомы 15 (15q11-q13), причем только в хромосоме, унаследованной от отца больного. Таким образом, геном таких пациентов имеет генетическую информацию в области 15q11-q13, происходящую только от матерей. И наоборот, примерно у 70% пациентов с редким синдромом Ангельмана, характеризующегося необычным лицом, низким ростом, выраженным интеллектуальным отставанием, спастикой и судорогами, отмечают делецию приблизительно той же хромосомной области, но теперь в хромосоме, унаследованной от матери; т.е. пациенты с синдромом Ангельмана имеют генетическую информацию в регионе 15q11-q13, происходящую только от отцов. Эта необычная ситуация удивительным образом доказывает, что родительское происхождение генетического материала в описанных случаях (в хромосоме 15) имеет выраженное влияние на клиническое проявление дефекта.
Приблизительно 30% пациентов с синдромом Прадера-Вилли не имеют цитогенетически обнаруживаемых делеций; но у них выявлены две цитогенетически нормальные хромосомы 15, обе унаследованные от матери. Ситуация иллюстрирует однородительскую дисомию — наличие дисомной линии клеток, содержащих две хромосомы или их части, унаследованные от одного родителя. Если оба экземпляра представлены идентичной хромосомой, состояние называют изодисомией; если присутствуют разные гомологи от одного родителя — гетеродисомией. Приблизительно 3-5% пациентов с синдромом Ангельмана также имеют однородительскую дисомию, только с двумя неповрежденными хромосомами 15 отцовского происхождения. Эти пациенты служат дополнительным подтверждением того, что синдромы Прадера-Вилли и Ангельмана — результат потери соответственно отцовского или материнского вклада генов участка 15q11-q13. Кроме хромосомной делеции и однородительской дисомии, несколько пациентов с синдромами Прадера-Вилли и Ангельмана, вероятно, имеют дефект в самом центре импринтинга. В результате не происходит переключения от женского к мужскому импринтингу в сперматогенезе или от мужского к женскому в овогенезе.
Оплодотворение сперматозоидом, несущим аномально персистирующий женский импринтинг, приведет к рождению ребенка с синдромом Прадера-Вилли; оплодотворение яйцеклетки, имеющей несвойственный ей мужской импринтинг, закончится рождением ребенка с синдромом Ангельмана. Наконец, мутации в материнской копии одного гена — убиквитин-протеин лигазы Е6-АР, как оказалось, вызывают синдром Ангельмана. Ген убиквитин-протеин лигазы Е6-АР расположен в области 15q11-q13 и в норме импринтирован, экспрессируется только материнский аллель в центральной нервной системе (ЦНС). Полагают, что крупные материнские делеции области 15q11-q13 и отцовские однородительские дисомии хромосомы 15, наблюдаемые при синдроме Ангельмана, служат причиной заболевания, так как приводят к утрате материнской копии критически важного импринтированого гена. Мутации для одного импринтированного гена при синдроме Прадера-Вилли пока еще не обнаружены. — Вернуться в содержание раздела «генетика» на нашем сайте Оглавление темы «Аномалии хромосом»:
|

Источник
Синдром Вольфа — Хиршорна. Синдром делеции длинного плеча хромосомы DСимптомокомплекс, обусловленный нехваткой дозы генов, локализованных в коротком плече 4-й хромосомы, описан в 1965 г. независимо О. Н. Wolff с соавторами и К. Hirschorn с соавторами. Частота его в популяции не установлена. Дети обычно рождаются с малой массой при доношенной беременности, отстают в физическом и психическом развитии. Безусловные рефлексы угнетены. Мышечный тонус снижен. Комплекс аномалий включает микроцефалию, гипертелоризм, маленькие низко расположенные уши, широкий уплощенный нос, расщепление губы и аномалии нёба. Менее постоянно могут быть: асимметрия черепа, крипторхизм, гипоспадия, паховые грыжи, клинодактилия, косолапость, избыток кожи на шее, дисплазия тазобедренных суставов, пороки сердца и внутренних органов. При цитогенетической верификации синдрома выявляется либо простая делеция короткого плеча 4-й хромосомы (4 р—), либо делеция, замаскированная транслоцированным фрагментом (см. синдром Эдвардса), либо кольцевая хромосома 4.
Синдром делеции длинного плеча хромосомы DСимптомокомплекс, обусловленный нехваткой генов, локализованных в длинном плече D-хромосомы, описан в 1969 г. К. Allderadice и др. Его частота в популяции не установлена. Клинически проявляется врожденной гипотрофией, задержкой психического и физического развития, микроцефалией, тригоноцефалией, микрогнатией, большими деформированными ушами, гипертелоризмом, выбухающим переносьем, микрофтальмией и птозом. Реже описывают асимметрию лица, выступающие вперед зубы и крыловидные складки на шее, различные дефекты строения глаз и расщепление нёба, аплазию и гипоплазию больших пальцев рук, пороки сердца, неперфорированное анальное отверстие. В ряде случаев у «этих больных развивается ретинобластома. При цитогенетическом исследовании обычно находят укорочение длинного плеча одной из D-хромосом (Dq) или кольцевую D-хромосому. Следует отметить, что вариабельность клинических проявлений этого синдрома частично связана с тем, что в одних случаях имеет место нехватка генов 13-й хромосомы, в других — 14-й. Принято считать, что, например, ретинобластома развивается только у больных с делецией хромосомы 14-й. Синдром делеции короткого плеча 18-й хромосомы. Симптомокомплекс, обусловленный нехваткой генов, локализованных в коротком плече 18-й хромосомы. Описан в 1963 г. I. De Grouchy и др. Синдром включает задержку психического развития, гипотрофию, низкорослость, эпикант, птоз, оттопыренные уши, седловидный нос, «карпий рот», кариес зубов. Менее постоянно описывают шейные крыловидные складки и расщелину лица, мышечную гипотонию, алопецию, малые аномалии пальцев рук и ног — синдактилию, микромелию. Нарушено предречевое и речевое развитие. Больные могут жить длительно. Среди больных преобладают женщины. Патологоанатомически определяется аринэнцефалия и отсутствие мозолистого тела. — Также рекомендуем «Синдром делеции 18-й хромосомы. Синдром кошачьих глаз — синдром Шмида—Фраккаро» Оглавление темы «Наследственные синдромы в неврологии»: |

Источник
Влияние хромосомных мутаций (кариотипа) на течение и прогноз хронического лимфолейкоза (ХЛЛ)Хромосомные аберрации, числовые и структурные, при использовании метода FISH обнаруживаются у 80 % больных хроническим лимфолейкозом (ХЛЛ). В 15—40 % случаев на протяжении болезни наблюдается эволюция кариотипа с появлением хромосомных аберраций или присоединением новых к уже существующим. Если основными хромосомными изменениями при неходжкинских лимфомах являются транслокации, то при хроничеких лимфолейкозах транслокации практически не встречаются, а самыми частыми нарушениями являются делеции. Наиболее часто (более чем у 55 % больных) обнаруживается делеция длинного плеча хромосомы 13 — 13q14, почти у 20 % больных — делеция длинного плеча хромосомы 11 — 11q22—23, у 7—8 % больных выявляется делеция короткого плеча хромосомы 17-17р13 и у 5—6% больных — делеция длинного плеча хромосомы 6 — 6q21. Много реже, чем потери генетического материала (делеции), обнаруживаются его приобретения. Наиболее частыми являются трисомия хромосомы 12 или увеличение ее длинного плеча (у 15—20 % больных). Как редкие находки описаны делеции 5q, 6p, 9q, 10q, 14q и трисомии хромосом 8 и 3 (всего у 3—5 % больных). У отдельных больных, чаще моложе 50 лет, выявляют сложные хромосомные аберрации, вовлекающие три хромосомы и более. Сопоставление найденных хромосомных аберраций с течением болезни установило отчетливую корреляцию между ними. Анализ кариотипа, клинических проявлений и длительности заболевания у 325 больных показал, что при изолированной делеции 13q наблюдается стабильное состояние или очень медленное прогрессирование с хорошим ответом на терапию (медиана выживаемости 133 мес — такая же, как у больных без хромосомных нарушений). Обнаружение трисомии хромосомы 12, 11q- и 17р- ассоциировано с неблагоприятным течением болезни. Медиана выживаемости больных с трисомией 12 равнялась 114 мес, с делецией 11q — 79 мес, а с делецией 17р —всего 32 мес. Сопоставление выявленных хромосомных аберраций с мутационным статусом у 340 больных показало, что хромосомные аберрации встречаются практически с одинаковой частотой: в группе с мутациями IgVH-генов у 77 %, без мутаций — у 76 % больных. Однако хромосомные аберрации, ассоциированные с благоприятным прогнозом (13q-), достоверно чаще определяются у больных с мутациями IgVH-генов (р=0,003), в то время как 11q- и 17р- — с высокой степенью достоверности при отсутствии мутаций (р=0,002). Эти данные подтверждены работами других исследовательских групп, показавших, что время до прогрессирования болезни достоверно короче у больных с 11q- и 17р-, особенно у больных с этими аберрациями и отсутствием мутаций IgVH-генов, чем у больных с 13q- и трисомией хромосомы 12. В группе больных с аберрацией 6q- было самое короткое время до прогрессирования болезни.
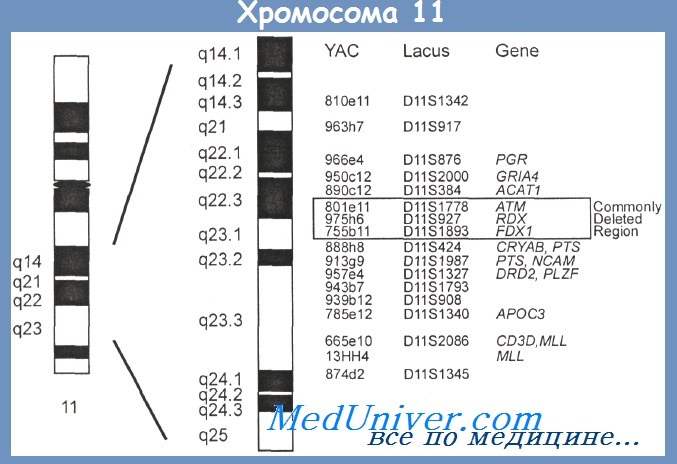
Итальянская кооперативная группа проанализировала методом FISH кариотип 217 больных хроническим лимфолейкозом (ХЛЛ). Делеция 6q21 обнаружена у 13 больных. Исследование мутационного статуса показало примерно равную частоту случаев с мутациями и без мутаций IgVH-генов, но клинически группа с делецией 6q21 была достаточно однородной: у всех больных отмечался высокий лейкоцитоз, у большинства — увеличение селезенки, у многих — атипичная морфология лимфоцитов несмотря на типичный для хронического лимфолейкоза (ХЛЛ) иммунофенотип. Всем больным лечение потребовалось сразу или вскоре после установления диагноза. Продолжительность жизни в этой группе была достоверно короче, чем у больных без данной хромосомной аберрации. Недавно при хроническом лимфолейкозе (ХЛЛ) обнаружена не описанная ранее t(1;6)(p35.3;p25.2). Как подчеркивалось ранее, транслокации не характерны для ХЛЛ. Однако указанная аберрация была обнаружена у 8 больных с типичными для ХЛЛ гематологическими и иммунологическими характеристиками. У 3 больных эта транслокация была единственной хромосомной аберрацией, у остальных сочеталась с характерными для ХЛЛ цитогенетическими изменениями: трисомией хромосомы 12, делециями 11q или 17р. Исследование мутационного статуса показало, что во всех случаях с t(1;6) не было мутаций IgVH-генов. Как известно, делеции часто вовлекают гены, которые являются супрессорами опухолевого роста. Если имеется делеция одного аллеля и мутация, даже точечная, другого, происходит функциональная инактивация соответствующего гена. В области 13ql4 — месте наиболее частой делеции при хроническом лимфолейкозе локализуется ген ретинобластомы RB1, который кодирует фосфопротеин, участвующий в регуляции транскрипции и контроле клеточного цикла. Моноаллельная делеция гена RB1 часто обнаруживается при хроническом лимфолейкозе, но инактивация гена в результате поражения второго аллеля является редкостью. Исследование нескольких других генов, идентифицированных в районе 13q, также не обнаружили при хроническом лимфолейкозе биаллельного выключения. Второй по частоте делецией при хроническом лимфолейкозе является 11q. Наиболее часто делегированным оказывается район 11q22.3—23.1. В этом районе локализованы два гена: АТМ (ataxia telangiectasia mutated) и RDX(radixin), гомолог гена нейрофиброматоза типа 2. Ген ATM кодирует образование белка, имеющего функцию протеинкиназы, участвующей в репарации ДНК и контроле клеточного цикла. Как известно, при атаксии-телеангиэктазии — заболевании, при котором имеется биаллельное изменение гена ATM, наблюдается повышенная частота лимфопролиферативных заболеваний. В больших сериях исследований при хроническом лимфолейкозе не найдено биаллельных изменений указанных генов. Тем не менее, многие наблюдения подтверждают, что аберрации 11q ассоциированы с выраженной лимфаденопатией, часто с увеличением лимфатических узлов в брюшной полости, а иногда и медиастинальных, ранним прогрессированием болезни с быстро наступающей потребностью в терапии и короткой продолжительностью жизни. Обнаруженный низкий уровень экспрессии ряда молекул адгезии, возможно, объясняет более быстрое распространение опухоли при делеции 11q. Н. Dohner и соавт. показали, что наиболее плохой прогноз у больных моложе 55 лет с делецией длинного плеча хромосомы 11: в этой группе медиана выживаемости составила 64 мес, в то время как у больных моложе 55 лет без делеции 11q — 209 мес. В более старшей возрастной группе продолжительность жизни достоверно не различалась — 94 мес и 111 мес для больных с делецией и без делеции 11q соответственно. При высокодозной терапии и последующей аутотрансплантации в случаях с делецией 11q23 достоверно чаще наблюдалось персистирование патологических клеток: при повторных исследованиях в течение 12 мес они постоянно определялись у 38 % больных с делецией 1 lq23 и только у 6 % (р = 0,014) больных без хромосомных аберраций или с другими аберрациями. Изучение экспрессии генов с помощью ДНК-микрочипов обнаружило 78 генов, по экспрессии которых случаи с делецией 1 lq23 достоверно отличались от остальных. До сих пор не удалось точно установить, какой сегмент удваивается при увеличении длинного плеча хромосомы 12; в разных исследованиях получены данные о вовлечении районов 12q13, 12q14, 12q15. Некоторые авторы отмечали нередкую атипичную морфологию лимфоцитов у больных с трисомией хромосомы 12 Разные авторы приводят противоречивые данные о прогностической роли трисомии хромосомы 12, но продолжительность жизни во всех исследованиях оказывается хуже, чем у больных с нормальным кариотипом. При трисомии хромосомы 12 нередко обнаруживается экспрессия Ki 67 — иммунологического маркера повышенной пролиферативной активности. Экспрессия этого маркера при хроническом лимфолейкозе нередко коррелирует с продвинутой стадией болезни. При использовании метода FISH установлено, что трисомия хромосомы 12 нередко сочетается с другими хромосомными аберрациями: делециями длинного плеча хромосом 13 и 14, трисомией хромосом 18 и 19. В отдельных случаях с течением времени трисомия хромосомы 12 появляется как вторая аберрация у больных с другими хромосомными нарушениями. Исследование мутационного статуса обнаружило признаки мутаций IgVH-генов у всех больных с трисомией 12 и отсутствие мутаций почти у всех больных с изменениями хромосомы 14. Анализ продолжительности жизни в зависимости от мутационного статуса и наличия различных изменений кариотипа установил, что продолжительность жизни при изолированной трисомии хромосомы 12 (108 мес) достоверно не отличается от продолжительности жизни больных с комплексными хромосомными аберрациями (89 мес; р = 0,612). В данном исследовании у всех больных с трисомией 12 были признаки мутаций генов IgVH, однако продолжительность жизни в этой группе достоверно не отличалась от продолжительности жизни больных без мутаций (95мес). В другом исследовании из этой же лаборатории на большой группе больных была показана отчетливая корреляция между наличием трисомии хромосомы 12 и отсутствием мутаций IgVH-генов при агрессивном течении хронического лимфолейкоза и наличием мутаций и структурными аномалиями района 13q14 при индолентном течении болезни. — Вернуться в раздел «гематология» Оглавление темы «Хронический лимфолейкоз (ХЛЛ)»:
|
Источник