Синдром дефекта ногтей и коленной чашечки

Синдром ногтей-надколенника
Синдром ногтей-надколенника (синдром nail-patella, NPS, остеониходисплазия, МКБ-10
— Q87.2) — редкое наследственное заболевание, которое характеризуется сочетанием
дисплазии ногтей, патологии костной системы и почек. Заболевание дебютирует в
младенчестве или детском возрасте. Лечение носит симптоматический характер. Прогноз
для пациентов определяется степенью поражения почек.
Синдром ногтей-надколенника связан с мутациями гена LMX1B.
Первое упоминание о больном с дистрофией ногтей и дисплазией скелета было сделано Э.
Шателайном (E. Chatelaine) в 1820 г. [1]. В 1930 г. У. Остеррайхер (W. Oesterreicher)
охарактеризовал триаду клинических симптомов синдрома ногтей-надколенника,
включающую в себя атрофию ногтей, аплазию надколенников и вывих головки лучевой
кости [2]. В 1950 г. К. Ф. Хокинс (C. F. Hawkins) и О. Е. Смит (O. E. Smith) установили,
что поражение почек является неотъемлемой частью данного синдрома [3].
Ген LMX1B, связанный с развитием синдрома ногтей-надколенника, был картирован в
1997 г. [4].
Распространенность и тип наследования
Заболевание дебютирует в младенчестве или детском возрасте. В 95 % случаев
врожденная патология ногтей при синдроме ногтей-надколенника проявляется в виде
гипоплазии или дисплазии ногтевых пластин (треугольные лунки, продольная
исчерченность, утолщение, губчатость) и в виде отсутствия ногтевой пластины. В 92 %
случаев выявляется гипоплазия и дисплазия надколенной чашечки, реже — ее отсутствие.
Нередко патология надколенника сопровождается недоразвитием латерального
надмыщелка бедренной кости, что ведет к рецидивирующим подвывихам надколенника. В
70 % случаев у пациентов имеются костные образования на внутренней поверхности
гребней подвздошных костей, так называемые «подвздошные рога». У многих больных
отмечается дисплазия костей предплечья в сочетании с гипоплазией головок плечевой и
лучевой костей, что приводит к повторным подвывихам локтевых суставов. У
большинства пациентов имеется дефицит массы тела независимо от характера питания,
наблюдаются поражения почек в виде аномалий развития органов мочевой системы,
протеинурии (присутствие белка в моче), изолированной или в сочетании с гематурией
(присутствие крови в моче). В 25 % случаев наблюдается нефропатия (поражение
клубочкового аппарата и паренхимы почек; у пациентов с данным синдромом —
аномалия гломерулярной базальной мембраны клубочков), в 10 % случаев развивается
хроническая почечная недостаточность. У пациентов встречаются варусная или
вальгусная деформации стоп, нарушения слуха, патологии органов зрения в виде
изменений роговицы (микрокорнеа — уменьшение размеров или уплощение роговицы,
склерокорнеа — частичная или полная «склеризация» роговицы (замещение ткани
роговицы склерой)), краевой пигментации радужки, врожденных катаракты и глаукомы
[7].
Первичную диагностику заболевания проводит педиатр. Диагноз ставится на основании
клинической картины, рентгенографии скелета, УЗИ почек, клинического анализа мочи,
консультации офтальмолога и отоларинголога. Диагноз подтверждается с помощью
молекулярно-генетического анализа LMX1B. Для данного заболевания возможна
пренатальная диагностика на основании биопсии ворсин хориона или амниоцентеза с
последующим молекулярно-генетическим анализом гена LMX1B [7].
В настоящее время специфическое лечение синдрома отсутствует, применяется
симптоматическая терапия. Для лечения и профилактики почечной недостаточности
применяются ингибиторы АПФ (ангиотензинпревращающего фермента). В терминальной
стадии хронической почечной недостаточности рекомендуется трансплантация почек [8].
Прогноз для пациентов определяется степенью поражения почек. Поражение ногтей при
синдроме ногтей-надколенника сохраняется на всю жизнь [8].
Синдром ногтей-надколенника связан с мутациями гена LMX1B [9]. Ген LMX1B кодирует
фактор транскрипции, играющий важную роль в развитии конечностей и почек. Белок
LMX1B регулирует экспрессию генов, кодирующих такие белки, как нефрин, подоцин,
CD2AP, а также цепи коллагена α3(IV) и α4(IV). Мутации гена приводят к нарушению
синтеза коллагена (основного структурного компонента гломерулярной базальной
мембраны почек) и дефекту подоцина, что связано с аномалиями ногтей и нарушением
функционирования почек [10].
- Ген LMX1B: c.176G>T (p.Cys59Phe)
- Ген LMX1B: c.305A>G (p.Tyr102Cys)
- Ген LMX1B c.306C>G (p.Tyr102Ter)
- Chatelaine (1820), quote by Roeckerath. W. Fortschritte auf der Gebiete der Rontgenstrahlen
1951: 75, 700–4. - Oesterreicher, W. Gemeinsame Vererbung von Anonychie bzw. Onychatrophie,
Patellardefekt und Luxatio radii. Dominantes Auftreten in 5 Generationen. Ztschr.
Konstitutionslehre 15: 465-476, 1930. - Hawkins, C. F., Smith, O. E. Renal dysplasia in a family with multiple hereditary
abnormalities including iliac horns. Lancet 255: 803-808, 1950. Note: Originally Volume I
[PubMed: 15416035] - Iannotti, C. A., Inoue, H., Bernal, E., Aoki, M., Liu, l., Donis-Keller, H., German, M. S.,
Permutt, M. A. Identification of a human LMX1 (LMX1.1)-related gene, LMX1.2: tissue-
specific expression and linkage mapping on chromosome 9. Genomics 46: 520-524, 1997
[PubMed: 9441763] - Sweeney, E., Fryer, A., Mountford, R., Green, A., McIntosh, I. Nail patella syndrome: a
review of the phenotype aided by developmental biology. J. Med. Genet. 40: 153-162, 2003
[PubMed: 12624132] - Renwick, J. H. Nail-patella syndrome: evidence for modification by alleles at the main locus.
Ann. Hum. Genet. 21: 159-169, 1956 [PubMed: 13373182] - Дружинина Т.В., Никонова О.П., Барсукова Т.И. Случай диагностики синдрома nail-
patella // Вестник Смоленской медицинской академии – 2010. — №4. – С. 78 – 81 - Константинова О.Д., Архипов В.В., Соловьев А.А., Майзельс И.Г.
Гормончувствительный вариант нефротического синдрома у ребенка с наследственной
остео-ониходисплазией // Нефрология – 2005. – Т.9. -№2. – С. 123 – 126 - Dreyer, S. D., Zhou, G., Baldini, A., Winterpacht, A., Zabel, B., Cole, W., Johnson, R. L.,
Lee, B. Mutations in LMX1B cause abnormal skeletal patterning and renal dysplasia in nail
patella syndrome. Nature Genet. 19: 47-50, 1998 [PubMed: 9590287] - Vollrath, D., Jaramillo-Babb, V. L., Clough, M. V., McIntosh, I., Scott, K. M., Lichter, P. R.,
Richards, J. E. Loss-of-function mutations in the LIM-homeodomain gene, LMX1B, in nail-
patella syndrome. Hum. Molec. Genet. 7: 1091-1098, 1998. Erratum: Hum. Molec. Genet. 7:
1333 only, 1998 [PubMed: 9618165]
Источник
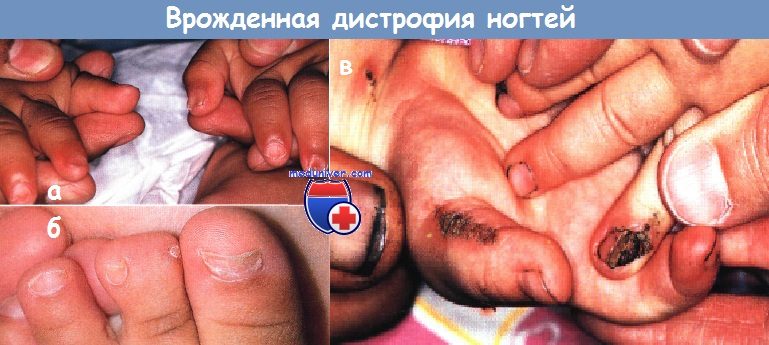
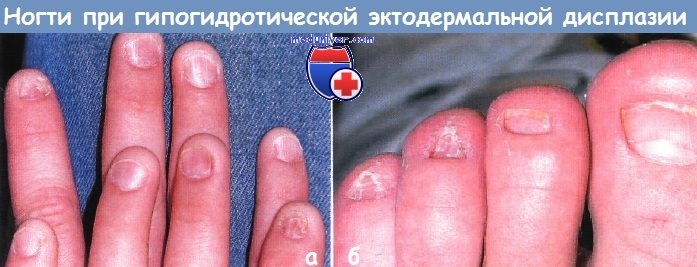
Врожденные и наследственные заболевания с нарушением роста ногтейОтсутствие, гипоплазия или дисплазия ногтей могут быть изолированными патологическими состояниями или же являться частью эктодермальной дисплазии или других генодерматозов. Аномалии ногтей: I. Анонихия: II. Наследственная эктодермальная дисплазия. III. Хромосомные аномалии: IV. Гиперпластические ногти: V. Койлонихия (ложкообразные ногти): VI. Медикаментозные мальформации: VII. Другие заболевания с дистрофией ногтей:
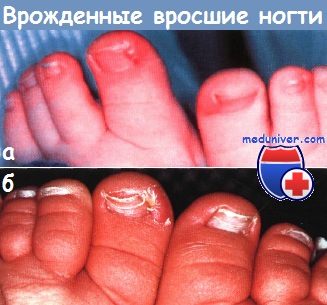
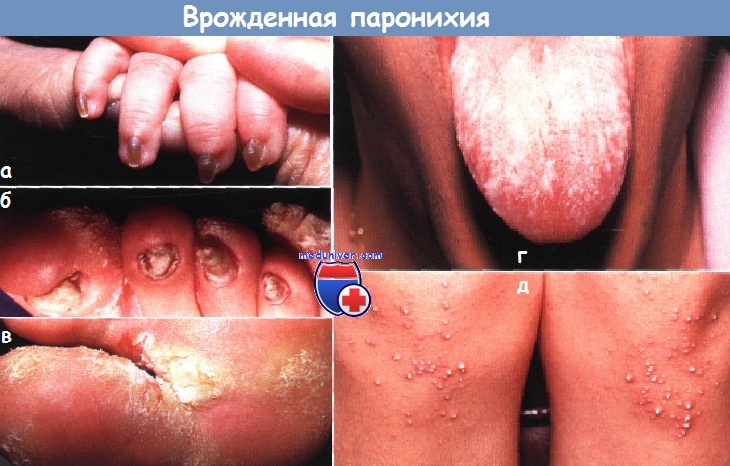
Поражения ногтей при изолированных мальформацияхАномальные ногти, изогнутые по типу часовых стекол (на пальцах типа «барабанные палочки»), и ложкообразные ногти могут быть наследуемыми по аутосомно-доминантному типу аномалиями без других сочетанных отклонений. Хотя врожденные вросшие ногти па пальцах стоп могут быть следствием врожденного смещения больших пальцев, которое корректируется только хирургическим путем, в большинстве случаев эти аномалии разрешаются самостоятельно и не связаны с анатомическим дефектом. Спонтанно регрессирующий вросший ноготь может быть результатом транзиторной гипоплазии ногтей стоп (особенно больших пальцев), которая обычно разрешается в пределах 12-18 мес. Изменения ногтей могут усиливаться вследствие внешней травмы и рецидивирующей/ хронической паронихии.
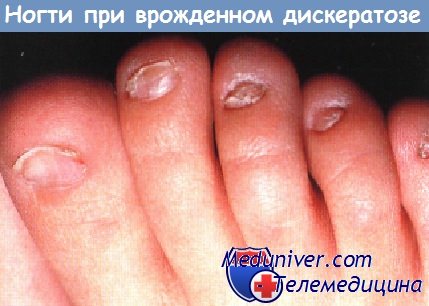
Поражения ногтей при эктодермальных дисплазияхМногие из эктодермальных дисплазии связаны с аномалиями ногтей; для некоторых из них они являются характерными и диагностически значимыми. При врожденной пахонихии, аутосомно-доминантной патологии с вариабельной пенетрацией, в первые несколько месяцев жизни развивается гиперкератоз ногтевого ложа. Затем происходит утолщение и удлинение ногтевой пластинки, приобретающей желтовато-коричневый цвет. Незначительная травма может привести к болезненному отделению ногтевой пластинки от ногтевого ложа и к кровотечению. Типичен гипергидроз ладоней и подошв, а также образование пузырей и мозолей. К другим признакам относятся лейкокератоз слизистой полости рта (не связанный со злокачественной дегенерацией), утолщение барабанных перепонок, приводящее к глухоте, леикокератоз роговицы и катаракты. Сообщалось также о гиперкератотических папулах на разгибательных поверхностях конечностей, дермоидных кистах и множественной стеатоцитоме. Врожденная пахонихия типа Ядассона-Левандовского, не связанная с глазными симптомами, вызывается мутацией в гене кератина 16 (KRT16) или в гене кератина 6А (KRT6A), расположенных в локусах 17q 12-21, 12q13. Дефекты в гене кератина 17 (KRT17) и кератина 6В (KRT6B) связаны с пахонихией по типу Джексона-Лоулера, при которой обнаруживается дистрофия сетчатки без лейкоплакии слизистой полости рта. Врожденный дискератоз можно ошибочно принять за врожденную пахонихию. Однако при врожденном дискератозе, который может быть сцепленным с Х-хромосомой, с аутосомно-доминантным или аутосомно-рецессивным типом наследования, ногтевая пластинка истонченная, наблюдаются продольные бороздки и птеригий. При врожденном дискератозе на шее и туловище выражена пойкилодермия с сетчатой пигментацией, телеангиэктазиями и атрофией. Лейкоплакия слизистой полости рта может ассоциироваться с развитием плоскоклеточного рака и другими злокачественными новообразованиями в ЖКТ. У 50% пациентов во второй и третьей декадах жизни отмечается панцитопения, которая напоминает анемию Фанкони. В большинстве случаев тип наследования рецессивный, сцепленный с Х-хромосомой, и ассоциируется с мутациями в локусе Xq28, в гене, кодирующем дискерин (DKC1). Гипоплазия ногтей может также быть признаком гидротической эктодермальной дисплазии и синдрома Коффина-Сириса.
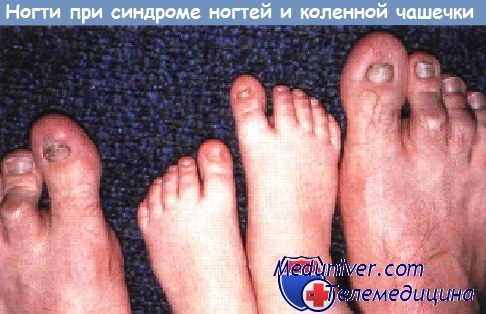
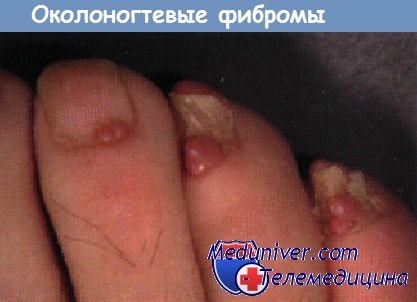
Поражения ногтей при генодерматозах и системных заболеванияхСиндром дефекта ногтей и коленной чашечки, аутосомно-доминантная аномалия, проявляется врожденным отсутствием или гипоплазией ногтей и коленной чашечки. В небольшом проценте случаев наблюдается гломерулонефрит, в редких случаях фатальный. Сообщалось также о гетерохромии радужной оболочки глаза, кератоконусе и катарактах. Околоногтевые фибромы, возникающие из проксимальной ногтевой бороздки, часто наблюдаются у пациентов с туберозным склерозом. Хотя фибромы появляются только в позднем детском или во взрослом возрасте, они могут стать первым ключом к диагнозу у лиц без других выраженных признаков поражения. Врожденная гипоплазия ногтей у детей, матери которых во время беременности принимали противосудорожные препараты, варфарин и другие тератогенные лекарственные средства, указывает на необходимость тщательной оценки ребенка на наличие других медикаментозно индуцированных признаков.
— Также рекомендуем «Паронихия у детей» Оглавление темы «Детская дерматология»:
|
Источник
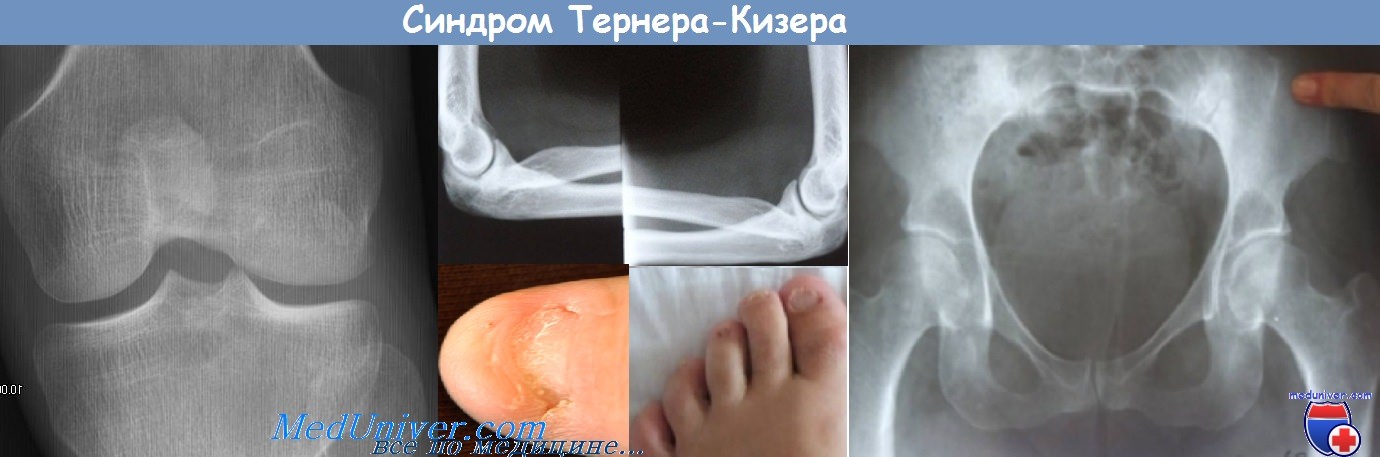
Синдром ногтя-надколенника (остеоониходисплазия) — клиника, диагностикаДля этого аутосомно-доминантного заболевания характерны гипоплазия или отсутствие надколенника, ониходистрофия, дисплазия локтевых суставов, экзостозы крыла подвздошной кости и поражение почек. Установлено, что ген синдрома ногтя—надколенника находится на длинном плече 9-й хромосомы (сегмент 9q34). У мышей инактивация гена LMX1B, кодирующего фактор транскрипции гомеобокса LIM, приводила к аномалиям скелета (гипоплазия когтей и отсутствие надколенника) и дисплазии почек. Оказалось, что человеческий гомолог этого гена, мутации в котором были обнаружены при синдроме ногтя—надколенника, находится именно в сегменте 9q34. Считается, что ген LMX1B важен для нормального развития конечностей и почек, но точные механизмы влияния его мутаций на поражение почек остаются неясными. Клинические признаки поражения почек есть менее чем у половины больных. Прогноз благоприятный, лишь у 10% больных с поражением почек развивается терминальная почечная недостаточность. Клинические проявления: микрогематурия и легкая протеинурия, возникающие в подростковом или юношеском возрасте. У некоторых больных развивается нефротический синдром. Если возникает артериальная гипертония, то, как правило, легкая. При световой микроскопии и иммунофлюоресценции специфических изменений в почках нет, но их можно выявить при электронной микроскопии. В базальной мембране клубочка находят множественные просветления неправильной формы, как будто она изъедена молью. При специальном окрашивании в этих просветлениях обнаруживают фибриллы с характерной для коллагена исчерченностью. Эти фибриллы могут встречаться в базальной мембране клубочка у больныхс синдромом ногтя—надколенника даже в отсутствие клинических проявлений поражения почек, но в других базальных мембранах их нет. Данных о природе этих фибрилл нет. Исчерченные фибриллы, оказавшиеся коллагеном III типа, были обнаружены в базальной мембране клубочка у больных с изолированным поражением почек, без ониходистрофии или костных аномалий, в ряде случаев эта так называемая болезнь коллагена III типа носила семейный характер. Большинство заболевших — дети, у них быстро развивается терминальная почечная недостаточность. В отличие от синдрома ногтя—надколенника болезнь коллагена III типа наследуется аутосомно-рецессивно, что указывает на то, что это разные болезни. Специального лечения поражения почек при синдроме ногтя—надколенника нет. Трансплантация почки дает хорошие результаты, без рецидивов в трансплантате. Поскольку заболевание наследуется аутосомно-доминантно, необходимо проверять потенциальных доноров из числа родственников на внепочечные проявления болезни.
— Также рекомендуем «Кистозные болезни почек — типы, диагностика, лечение» Оглавление темы «Наследственные болезни почек»:
|
Источник
Совместное (или сцепленное) наследование генов, находящихся в одной хромосоме, известно как закон сцепления Т. Моргана. Гены, расположенные в одной хромосоме, составляют группу сцепления. Число групп сцепления равно гаплоидному набору хромосом диплоидного организма. Например, у человека 23 группы сцепления, у дрозофилы — 4, у гороха — 7. Запись генотипов при сцепленном наследовании несколько видоизменяется. Генотип дигетерозиготы при независимом наследовании мы записываем как АаВв. При сцепленном наследовании этот же генотип записывается как авАВ или аВАв. Величина перекреста отражает расстояние между генами в хромосоме и измеряется как отношение кроссоверных особей к общему числу особей в потомстве от анализирующего скрещивания и выражается, в процентах. Единицей перекреста является одна морганида, или 1 % образования гамет, в которых произошел кроссинговер (сейчас используют термин). Сцепление генов может быть полное и неполное. Полное сцепление отмечается у самцов дрозофилы и самочек тутового шелкопряда. В этом случае в потомстве дигетерозиготы при анализирующем скрещивании будут возникать особи, имеющие только родительские сочетания в равном количественном отношении 1:1. При неполном сцеплении в F2 наряду с двумя фенотипическими классами с родительскими сочетаниями признаков в сравнительно небольшом числе будут возникать так называемые кроссоверные особи (особи, возникшие в результате сочетания кроссоверных гамет гибрида с гаметами рецессивной гомозиготы). Их появление связано с делением кроссинговера-обмена идентичными участками гомологичных хромосом в мейозе. Таким образом, образуется четыре фенотипических класса особей, но не в отношении 1:1:1:1, как при дигибридном скрещивании, а с преобладанием родительских классов над рекомбинантами. При решении задач нужно помнить, что особей родительских классов бывает более 50%, меньше всех бывает кроссоверов, возникших как результат двойного кроссинговера.
Задачи
1. Какие типы гамет и в каком численном соотношении образуются у родителей, имеющих генотип: АаВв, при независимом наследовании, при сцепленном и неполносцепленном наследовании
Решение
а) Дигетерозигота АаВв , согласно закону независимого наследования образует 4 типа гамет (АВ, Ав, аВ, ав) в равных численных отношениях (1:1:1.:1).
При полном сцеплении дигетерозигота АаВв продуцирует только 2 типа гамет (АВ и ав) в равных количественных отношениях. При неполном сцеплении дигетерозигота дает 4 типа гамет, из-за перекреста между генами А и В дополнительно образуется два типа кроссоверных гамет Ав и аВ, причем число их значительно меньше по сравнению с некроссоверными АВ и ав.
б) Гены сцеплены, кроссинговер между нами составляет 40%. Определите, сколько появится в потомстве дигетерозиготы авАВ при ее самооплодотворении форм аавв?
Решение.
Составим схему скрещивания:
P ♀АаВв x♂АаВв
Некроссоверные гаметы Некроссоверные гаметы
G AB ав AВ ав
Кроссоверные гаметы Кроссоверные гаметы
Ав aB Ав aВ
Оба родителя продуцируют по 4 типа гамет в соотношении 0,3; 0,3; 0,2; 0,2 исходя из величин кроссинговера (сумма некроссоверных равна 60%, или 0,6, а кроссоверных — 40%, или 0,4). По решетке Пеннета находим, что форм с генотипом аавв будет 0,09(0,3 ав х 0,3 ав), или 9%.
2. Организм имеет следующий генотип: АаВв. Расстояние между генами равно 15 морганидам. Напишите все типы гамет, которые образует организм с таким генотипом и укажите количество гамет каждого типа.
3. У человека локус резус-фактора сцеплен с локусом, определяющим форму эритроцитов, и находится от него на расстоянии 3 морганид. Резус-положительность и эллиптоцитоз (эритроциты эллиптической формы) определяются доминантными аутосомными генами. Один из супругов гетерозиготен по обоим признакам. При этом резус-положительность он унаследовал от одного родителя, эллиптоцитоз — от другого. Второй супруг резус-отрицателен и имеет нормальные эритроциты. Определите процентные соотношения вероятных генотипов и фенотипов детей в этой семье.
Решение
Условия задачи.
Признак Определяющий его ген
Резус-положительность Rh
Резус-отрицательность rh
Эллиптоцитоз А
Нормальная форма эритроцитов а
Генотип супруга, гетерозиготного по обоим признакам, RhrhАа. Генотип второго супруга rhrhaa. У первого супруга образуется 3% гамет с перекрестом (1,5% rha), остальные 97% — без перекреста (48,5% Rha и 48,5% rhA). Второй супруг дает гаметы только одного типа rha. Вероятность рождения детей с различными генотипами выразится в следующих отношениях: 48,5% Rhrhaa, 48,5%rhrhAa, 1,5% RhrhAa, 1,5% rhrhaa, то есть 48,5 резус-положительных с эритроцитами нормальной формой, 48,5% резус-отрицательных с эллиптоцитозом, 1,5 % резус-отрицательных с нормальными эритроцитами.
4. Синдром дефекта ногтей и коленной чашечки определяется доминантной аллелью гена. На расстоянии 10 морганид от него находится другой ген, определяющий группу крови по системе АВО. Один из супругов имеет II группу крови, другой – III. Тот, у которого II группа крови, страдает дефектом ногтей и коленной чашечки. Известно, что отец его был с I группой крови и не имел этих аномалий, а мать – с IV группой крови имела оба дефекта. Супруг, имеющий III группу крови, нормален в отношении гена дефекта ногтей и коленной чашечки и гомозиготен по обоим генам. Определите вероятность рождения в этой семье детей, страдающих дефектом ногтей и коленной чашечки, и возможные группы их крови.
5. Катаракта и полидактилия у человека обусловлены доминантными аутосомными тесно сцепленными генами( т. е. не обнаруживаемыми кроссинговера генами). Какое потомство можно ожидать в семье, где известно, что матеры обоих супругов страдали только катарактой, а отцы только полидактилией?
5. У человека врожденная глухота может определяться генами а и в. Для нормального слуха необходимо наличие в генотипе обеих доминантных аллелей (А-В-). Определите генотипы родителей в следующих двух семьях: а) оба родителя глухие, а их 7 детей имеют нормальный слух; б) у глухих родителей 4 глухих ребенка.
7. Одна из форм дефекта зубной эмали контролируется доминантным геном, расположенным в первой хромосоме, в этой же хромосоме расположен рецессивный ген расщелины верхней губы. Гены тесно сцеплены между собой. Что можно ожидать в потомстве: отсутствие аномалий, проявление двух или одной аномалии, если у отца присутствуют обе аномалии, а мать здорова и гомозиготна?
8. У человека тремы и нарушение прикуса наследуются сцепленно, как аутосомно-доминантные признаки. Гены данных аномалий расположены в 3 –й паре хромосом на расстоянии 12 морганид. Какова вероятность рождения детей с двумя аномалиями в семье, где отец здоров и гомозиготен, а мать имеет обе аномалии, одну из которых она получила от одного родителя, а другую – от другого?
9. В семье, где родители здоровы, родился ребенок с аномалией полости рта: расщелиной твердого нёба и гипоплазией гортани. Определите, как наследуются признаки, если гены тесно сцеплены и находятся в 22-й хромосоме. Какова вероятность рождения второго ребенка с этими аномалиями?
Источник