Синдром де груши у ребенка

В качестве примера синдрома со структурной перестройкой хромосом можно привести синдром делеции короткого плеча 5-й хромосомы — синдром 5р-, или синдром «кошачьего крика». Среди больных этим синдромом преобладают девочки. Частота встречаемости — 1:50 000 новорожденных. Характерные симптомы — это микроцефалия, круглое лицо (с возрастом оно вытягивается), широко расставленные глаза, антимонголоидный разрез глаз, эпикант, недоразвитие нижней челюсти, катаракта, косоглазие и другие глазные нарушения.
Синдром получил свое название в связи с тем, что крик новорожденных с этим синдромом напоминает кошачье мяуканье. Выраженная умственная отсталость отмечается во всех случаях. Среди детей с глубокой умственной отсталостью на эту патологию приходится 1% из всех случаев синдрома.
Трисомия-18
К синдромам с анеуплоидией относится трисомия-18, или синдром Эдвардса, встречающийся с частотой 1:7000. В данном случае лишней является хромосома 18-й пары. При этом синдроме преобладают девочки. Характерные симптомы синдрома — выступающий затылок, тонкие переносье и спинки 1, недоразвитие нижней челюсти, «птичий профиль», деформированные ушные раковины и тяжелые пороки развития внутренних органов (чаще всего сердца), в связи с чем такие дети умирают в раннем возрасте. До года доживают 10% детей с трсомией-18. Все больные отстают в умственном и физическом развитии.
Известны (описаны) и другие аномалии хромосомы-18, включающие как структурные, так и геномные перестройки.
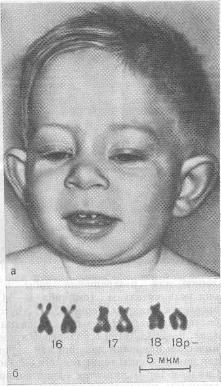
Синдром де Груши
Моносомия-18р. Частота встречаемости — 1: 60 000. Обычно ребенок рождается в срок, но с небольшой массой тела. В дальнейшем характерными признаками являются: маленький рост, микроцефальной формы череп, круглое лицо. Часто имеют место очаги облысения на голове либо тотальная алопеция. Характерны деформация зубов и ушных раковин, не редкость пупочных и паховых грыж на фоне мышечной гипотонии, аномалии кистей руки пальцев, синдактилия пальцев ног, «стопа-качалка». У мальчиков часто бывает недоразвитие половых органов, характерно резкое снижение продолжительности жизни у больных с грубой мозговой патологией. Реже наблюдаются более легкие формы интеллектуального дефекта с нормальной продолжительностью жизни. Характерно сочетание умственной отсталости с судорожным синдромом и различными речевыми расстройствами. Помимо делеции, возможны транслокационные и мозаичные варианты.
Синдром Лежена
Моносомия-18q, или синдром 18q — синдром Лежена. Делеция длинного плеча хромосомы-18. Встречается с частотой 1: 60 000. Девочки с этим синдромом рождаются в 1,5 раза чаще, чем мальчики. Одним из ранних характерных признаков считается синдром мышечной гипотонии: ребенок лежит на спине в «позе лягушки». Обращают на себя внимание микроцефальной формы череп, уплощенное лицо с выступающим подбородком. Характерна частота различных пороков развития зрительной системы, т.е. при данном синдроме имеет место дефект развития, включающий дефекты зрения и интеллекта. Среди нарушений зрительной функции преобладают колобомы, косоглазие, птоз, нистагм, снижение остроты зрения, атрофия зрительных нервов. Характерны своеобразная форма носа, рта, высокое твердое нёбо, иногда с расщелиной, своеобразная форма ушных раковин, нередко сужение или атрезия наружных слуховых проходов. Характерным признаком считается недоразвитие наружных половых органов, нередко отмечаются пороки сердца, почек. Интеллектуальные нарушения варьируются от легкой ^пограничной интеллектуальной недостаточности и даже нормального интеллекта) до олигофрении в степени идиотии.
Синдром Патау
К геномным мутациям относится также трисомия-13 — синдром Патау. Встречается синдром с частотой 1:6000 новорожденных. Дети рождаются с истинной пренатальной гипотрофией. В 50% случаев беременность осложняется многоводием. Типичным признаком является расщелина губы и неба. Как правило, дети страдают полидактилией. Характерны пороки развития органов зрения: катаракта, микрофтальмия, анофтальмия, циклопия. Череп неправильной формы, возможна тригоноцефалия, узкие глазные щели, запавшее переносье, деформированные низко расположенные ушные раковины, дефекты скальпа. Отмечаются рефлексорное сгибание костей, «стопа-качалка», пороки внутренних органов: врожденные пороки сердца, почек, желудочно-кишечного тракта, мочеполовой системы.
Продолжительность жизни резко снижена <Ф5% детей погибает в возрасте до года). В развитых странах отмечается тенденция к увеличению продолжительности жизни таких больных: 15% детей доживают до пятилетнего возраста и 2—3% — до десятилетнего возраста. Во всех случаях отмечается выраженное психическое недоразвитие.
Помимо трисомной (75%), встречаются транслокационная (20%) и мозаичная (5%) формы.
Синдром Реторе
С различными типами хромосомных аномалий может быть связана трисомия-9р — синдром Реторе. Предполагается, что в группе умственно отсталых детей трисомия по короткому плечу 9-й хромосомы занимает по частоте второе место после
рома Дауна. Девочки с данным синдромом встречаются в два раза чаще, чем мальчики. Череп у новорожденных микробранхицефальный с уплощенным затылком. С возрастом брахицефалия уменьшается. Роднички широко открыты, имеется лобный шов. Характерны глазные аномалии: микро- или энофтальмия, страбизм, нарушения рефракции, гипертелоризм эпикант, антимонголоидный разрез глаз, крупный нос с широким кончиком, опущенные углы рта, короткая верхняя губа, «конские» зубы, диспластичное телосложение. У 25% детей врожденные пороки сердца. Умственная отсталость диагносцируется у всех больных. Выраженность интеллектуального дефекта варьируется от легкой до глубокой; типичны эмоциональная лабильность, повышенная психомоторная возбудимость, двигательные расстройства с нарушением координации движений. Среди цитогенетических вариантов синдрома Реторе — транслокационный, связанный с частичной трисомией (иногда с тетрасомией), «свободная» трисомия, мозаицизм предполагается, что характерные фенотипические проявления связаны с сегментом 9р21.
Читайте также:
Рекомендуемые страницы:
©2015-2020 poisk-ru.ru
Все права принадлежать их авторам. Данный сайт не претендует на авторства, а предоставляет бесплатное использование.
Дата создания страницы: 2016-02-12
Нарушение авторских прав и Нарушение персональных данных
Поиск по сайту:
Источник
Причина.
Делеция короткого плеча 18-й хромосомы
(возможны транслокационные и мозаические
варианты).
Клиника.
Низкая масса тела при рождении, профиль
плоский или вогнутый. Маленький рост,
микроцефалитная форма черепа, круглое
лицо, высокое небо (иногда с расщелиной).
Очаги облысения на голове либо тотальная
алопеция. Характерна деформация зубов
и ушных раковин, пупочечные и паховые
грыжи. Аномалия кистей рук и пальцев,
синдактилия пальцев ног. У мальчиков
часто недоразвитие половых органов.
Отмечается
умственная отсталость.
Патогенез.
У больных с грубой мозговой патологией
резкое снижение продолжительности
жизни. Косоглазие, мышечная гипотония,
гипоплазия полового члена и мошонки у
мальчиков и гипоплазия малых половых
губ у девочек. Пороки сердца, иногда
почек.
Диагностика.
Исследование кариотипа, цитологическое
обследование.
Лечение.
Симптоматическое.
Частота.
1: 60000.
Синдром Лежена
Причина.
Делеция длинного плеча 18-й хромосомы.
Клиника.
Синдром мышечной гипотонии. Ребенок
лежит на спине в позе «лягушки». Череп
микроцефальной формы, уплощенное лицо
с выступающим подбородком. Косоглазие,
птоз, нистагм, снижение зрения, высокое
твердое небо (иногда с расщелиной),
своеобразная форма ушных раковин,
нередко сужение слуховых органов.
Интеллектуальное
нарушение варьируется от легкой до
олигофрении в степени идиотии.
Патогенез.
Пороки сердца, иногда почек. Пороки
развития зрительной системы.
Диагностика.
Дерматоглифика (повышенное число
завитков на пальцах рук), цитологическое
обследование.
Частота.
1:60000.
Синдром Реторе
Причина.
Частичная трисомия по короткому плечу
хромосомы 9.
Клиника.
Череп у новорожденных микробранхицефальный
с уплощенным затылком. С возрастом
брахицефалия уменьшается. Роднички
широко открыты, имеется лобный шов.
Характерны глазные аномалии: микро- или
энофтальмия, страбизм, нарушение
рефракции, эпикант, антимонголоидный
разрез глаз, крупный нос с широким
кончиком, опущены углы рта, короткая
верхняя губа, «конские» зубы, дипластичное
телосложение. Умственная
отсталость варьируется от легкой до
глубокой.
Патогенез.
Глазные аномалии, у 25%- врожденные пороки
сердца. Двигательные расстройства,
нарушение координации.
Диагностика.
Дерматоглифика, цитологическое
обследование, кариологическое
исследование.
Лечение.
Отсутствует.
Синдром Брадера – Вилли
Причина.
Утраивается участок 15-й хромосомы
отцовского происхождения
Клиника.
Мышечная гипотония, половое недоразвитие,
ожирение, умственная отсталость. Низкая
масса тела при рождении, пониженная
температура. Потом развивается
чрезвычайный аппетит. Деформированные
низко расположенные ушные раковина и
мягкие ушные хрящи, подковообразная
форма рта, короткая губа, неправильный
рост зубов. Диспропорциональность стоп
и кистей. Нарушение осанки.
Патогенез.
Частые грыжи, патология кистей и стоп.
В пубертатном возрасте диабет. Взрослые
страдают гиперсомнией, ишемической
болезнью сердца, инфаркт миокарда.
Диагностика.
Клиническое обследование.
Лечение.
Симптоматическое.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
Источник
Контрольная работа. Тема: Хромосомные болезни вызванные нарушением числа аутосом
скачать (273 kb.)
Доступные файлы (1):
содержание
- Смотрите также:
- Генетика пола. Закономерности наследования. Хромосомные болезни [ реферат ]
- Хромосомы и хромосомные болезни [ реферат ]
- Психолого-педагогические особенности детей с нарушением зрения [ лабораторная работа ]
- Неинфекционные болезни растений, вызванные дефицитом микроэлементов [ реферат ]
- Неинфекционные болезни растений [ документ ]
- Дипломная работа — Адаптивная физическая культура для детей с нарушением зрения [ дипломная работа ]
- Генные и хромосомные болезни человека [ лекция ]
- Хромосомные болезни пола (синдром Тернера, синдром трисомии X) [ реферат ]
- по Visual Basic [ документ ]
- адаптивная физическая культура для детей с нарушением зрения [ реферат ]
- Паспорта специальностей — Медицинские науки [ документ ]
- Физиотерапия язвенной болезни желудка и двенадцатиперстной кишки [ курсовая работа ]
1.doc
ГБОУ СПО НСО
«Новосибирский педагогический колледж им. А .С. Макаренко»
Контрольная работа
по основам генетики.
Мусиной Натальи Николаевны
Специальность 050710
«Специальное дошкольное
образование» Курс 3 ,
группа305
Форма обучения : заочная.
2011
Вариант№5.
Тема:
« Хромосомные болезни вызванные нарушением числа аутосом.»
План:
1. Понятие об аутосомах ,их количестве ,функциях.
2. Частичные и полные моносомии:
• синдром «кошачьего крика»
• синдром Лежена
• синдром де Груши
3 .Трисомии :
• синдром Дауна
• синдром Эдвардса
• синдром Патау
Все заболевания человека с учетом роли наследственных факторов можно разделить на три группы:
1.Наследственные болезни, которые развиваются только при наличии мутантного гена; они передаются из поколения в поколение через половые клетки; например, некоторые формы мышечной дистрофии, близорукость, шестипалость.
2.Болезни с наследственным предрасположением; в этом случае передаются не сами болезни, а предрасположенность к ним; для развития таких заболеваний необходимы дополнительные внешние вредные воздействия; например, эпилепсия, некоторые аллергические состояния, гипертоническая болезнь.
3.Заболевания, которые вызываются различными инфекционными агентами, обусловлены травмой и непосредственно как бы не зависят от наследственности. Однако и в этих случаях она играет определенную роль. Известно, что в некоторых семьях имеется несколько больных туберкулезом, в других – дети часто страдают простудными заболеваниями. Не все люди, имеющие контакт с инфекционными больными заболевают, и, наконец, нельзя исключить, что в многообразии течения заболевания определенную роль играют наследственные особенности организма.
Хромосомные болезни или хромосомные синдромы – это комплексы множественных врожденных пороков развития, вызываемых числовыми или структурными изменениями хромосом, видимыми в световой микроскоп.
Нарушения в строении хромосом, изменения их количества, генные мутации могут возникать на разных этапах развития организма. Если они возникают в гаметах родителей, то аномалия будет наблюдаться во всех клетках организма (полный мутант).
Если они возникают в процессе эмбрионального развития, хромосомный набор в разных клетках тела будет разным. В процессе развития появляется несколько следующих друг за другом поколений клеток с различными хромосомными наборами. При незначительном количестве аномальных клеток болезни в последующем может и не быть.
1.Понятие об аутосомах ,их количестве ,функциях.
Хромосомы состоят из 2 сестринских хроматид (удвоенных молекул ДНК), соединенных друг с другом в области первичной перетяжки — центромеры. Центромера делит хромосому на 2 плеча. В зависимости от расположения центромеры хромосомы бывают; 1) метацентрические центромера расположена в середине хромосомы и плечи ее равны; 2)субметацентрические центромера смещена от середины хромосом и одно плече короче другого; 3) акроцентрические — центромера расположена близко к концу хромосомы. И одно плечо значительно короче другого. В некоторых хромосомах есть вторичные перетяжки, отделяющие от плеча хромосомы участок, называемый спутником, из которого в интерфазном ядре образуется ядрышко,
Правила хромосом
1. Постоянство числа хромосом.
Соматические клетки организма каждого вида имеют строго определенное число хромосом (у человека -46)
2. Парность хромосом.
Каждая. хромосома в соматических клетках с диплоидным набором имеет такую же гомологичную (одинаковую) хромосому, идентичную по размерам, форме, но неодинаковую по происхождению: одну — от отца, другую — от матери.
3. Правило индивидуальности хромосом.
Каждая пара хромосом отличается от другой пары размерами, формой, чередованием светлых и темных полос.
4. Правило непрерывности.
Перед делением клетки ДНК удваивается и в результате получается 2 сестринские хроматиды. После деления в дочерние клетки попадает по одной хроматиде, таким о6разом, хромосомы непепрывны: от хромосомы образуется хромосома.
Все хромосомы подразделяются на аутосомы и половые хромосомы. Половые — это 23 пара хромосом, определяющая формирование мужского 11 женского организма.
Аутосомы — все хромосомы в клетках, за исключением половых хромосом, их 22 пары.
В соматических клетках присутствует.двойной — диплоидный набор хромосом, в половых-, гаплоидный (одинарный). Хромосомный набор здорового человека — 46 хромосом: 22 пары аутосом и 1 пара половых хромосом ( женщина — ХХ, мужчина — ХУ).
Определенный набор хромосом клетки, характеризующийся постоянством их числа, размером и формой, называется кариотипом .
Для того, чтобы разобраться в сложном наборе хромосом, их располагают попарно по мере убывания их величины, с учетом! положения центромеры и наличия вторичных перетяжек. Такой систематизированный кариотип называется идиограммой.
Для изучения кариотипа генетики используют метод цитогенетического анализа, при котором можно диагностировать ряд наследственных заболеваний, связанных с нарушением числа и формы хромосом.
Этиологическими факторами хромосомной патологии являются все виды хромосомных мутаций (хромосомные аберрации) и некоторые геномные мутации (изменения числа хромосом). У человека встречаются только 3 типа геномных мутаций: тетраплоидия, триплоидия и анеуплоидия. Из всех вариантов анеуплоидий встречаются только трисомии по аутосомам, полисомии по половым хромосомам (три-, тетра- и пентасомии), а из моносомий – только моносомия X.
У человека обнаружены все типы хромосомных мутаций: делеции, дупликации, инверсии и транслокации. Делеция (нехватка участка) в одной из гомологичных хромосом означает частичную моносомию по этому участку, а дупликация (удвоение участка) – частичную трисомию.
Унаследовавшего такие измененные хромосомы от одного из родителей, будет частичная моносомия по одному или двум концевым участкам хромосомы. Транслокации — перенос участка из одной хромосомы в другую или в другое место этой же хромосомы.
Исходы — гибель, ВПР, высокий риск рождения больных детей. Например, слияние 2-х хромосом в одну (синдром Дауна) — 21 хромосома с 14-ой или 15-ой.
Инверсия — при разрыве хромосомы в двух местах освобожденный участок разворачивается на 180% и вновь встает на прежнее место.
Исходы — спонтанные аборты, множественные ВПР, малые аномалии развития, умственная отсталость, без аномалий.
Делеция — исчезновение оторванной части хромосом. У каждой хромосомы выделяются длинное и короткое плечо. Короткое плечо обозначается маленькой латинской буквой «р», длинное плечо — «q». Недостаток одного какого-либо плеча хромосомы обозначается соответствующей латинской буквой, после чего ставится знак «-», а цифра, стоящая перед буквой указывает порядковый номер аномальной хромосомы.
Такие мутации сильно изменяют фенотип, т.к. изменяется много генов, не накапливаются в генофонде, т.к. у них очень высокая летальность. Хромосомные мутации могут так же быть материалом для естественного отбора и селекции.
2.Частичные и полные моносомии
Моносомия – это наличие всего одной из пары гомологичных хромосом. В случае обширной делеции в какой-либо хромосоме иногда говорят о частичной моносомии.
• Синдром кошачьего крика – частичная моносомия по короткому плечу аутосомы 5 р-). Критический регион у всех пациентов 5р15.2
Синдром кошачьего крика (5 р-) обусловлен делецией короткого плеча 5-й хромосомы. Популяционная частота синдрома – примерно 1:45 000 Фенотип включает внутриутробную задержку роста, микроцефалию, трудности при кормлении, врожденный порок сердца, врожденный вывих бедра, косолапость и тяжелую гипотонию. Лицевые черты в неонатальном периоде включают круглое лицо, опущенные углы глаз, гипертелоризм, диспластические, низкосидящие, ротированные назад уши, опущенные углы рта. Название синдром получил в связи с характерным писком, напоминающим кошачий, у пораженных детей, но это наблюдается не во всех случаях и может быть у детей с другими хромосомными аномалиями. Характерные лицевые черты и крик ребенка обращают на себя внимание, а диагноз может быть подтвержден рутинным хромосомным анализом и FISH-анализом с пробами для критического региона. Иногда наблюдаются атрофия зрительного нерва и очаги депигментации сетчатки. Как правило, выявляются пороки сердца. Наиболее постоянный признак синдрома – «кошачий крик» – обусловлен изменениями гортани: сужением, мягкостью хрящей, отечностью или необычной складчатостью слизистой оболочки, уменьшением надгортанника. Изменения других органов и систем неспецифичны.
Продолжительность жизни у больных с этим синдромом значительно снижена, только около 14% из них переживают возраст 10 лет.
• Синдром Лежена-делеция длинного плеча хромосомы-18 (медосомия-18g или синдром 18g).Встречается с частотой 1:60 000 .Девочки рождаются 1,5 раза чаще чем, мальчики .Одним из ранних характерных признаков считается синдром мышечной гипотонии: ребенок лежит на спине в «позе лягушки» . Обращают на себя внимание череп микроцефальной формы ,уплощенное лицо с выступающим подбородком .Характерна частота различных пороков развития зрительной системы (включается дефект зрения и интеллекта). Среди нарушений зрительных функций преобладают-коломбы, косоглазие ,птоз (опущение век)нистагм(дрожание глаз),снижение остроты зрения ,атрофия зрительных нервов. Характерны своеобразная форма носа, рта, высокое твердое небо иногда сужение или атрезия наружных слуховых проходов .Характерным признаком является недоразвитие наружних половых органов .Нередко отмечаются пороки сердца ,почек .Интелект-от легкой (пограничной )степени недоразвития (и даже нормальной степенью интелекта)до олигофрении .
• Синдром де Груши -моносомия 18p. Частота встреемости 1 :60 000 .обычно ребенок рождается в срок,но с небольшой массой тела.В дальнейшем харакерным признаком является маленький рост микроцефальной формы череп ,круглое лицо.Часто имеют место очаги облысения на голове или тотальная аллопеция .Характерны деформация зубов и ушных раковин, не редки пупочные и паховые грыжи (на фоне мышечной гипотонии),аномалии кистей рук и пальцев ,синдактилия пальцев ног, «стопа-качалка».У мальчиков часто бывает недорозвитие половых органов .Характерно снижение продолжительности жизни при грубых мозговых нарушениях и более легкие формы интелектуальных дефектов с нормальной продолжительностью жизни ,сочетание умственной отсталости с судорожным синдромом и различными речевыми расстройствами. Помимо делеции возможны мозаичные и транслокационные варианты.
3. Трисомии
Трисомия — хромосомная аномалия из-за нерасхождения хромосом, протекает тяжелее, чем аномалии половых хромосом. Моносомии по аутосомам не совместимы с жизнью.
1. Болезнь Дауна (синдром трисомии 21 пары, монголизм) (Q90.9)
Кариотип 47 ХХ или 47 ХУ, 21+. Соотношение полов — МI: ЖI. Частота — 1: 700-800.( слияние 2-х хромосом в одну 21 хромосома с 14-ой или 15-ой.)
В нашей стране ежегодно рождается около 8 тысяч детей с болезнью Дауна. Синдром Да́уна (трисомия по хромосоме 21) — одна из форм геномной патологии, при которой чаще всего кариотип представлен 47 хромосомами вместо нормальных 46, поскольку хромосомы 21-й пары, вместо нормальных двух, представлены тремя копиями (трисомия, см. также Плоидность). Существует ещё две формы данного синдрома: транслокация хромосомы 21 на другие хромосомы (чаще на 15, реже на 14, ещё реже на 21, 22 и Y-хромосому) — 4% случаев, и мозаичный вариант синдрома — 5%.
Синдром получил название в честь английского врача Джона Дауна (John Down), впервые описавшего его в 1866 году. Связь между происхождением врождённого синдрома и изменением количества хромосом была выявлена только в 1959 году французским генетиком Жеромом Леженом.
Слово «синдром» означает набор признаков или характерных черт. При употреблении этого термина предпочтительнее форма «синдром Дауна», а не «болезнь Дауна».
Клиника:
• — характерная внешность: небольшая круглая голова со скошенным утолщенным затылком: монголоидный разрез глаз, эпикант, короткий седловидный нос, маленькие отстающие деформированные ушные раковины, полуоткрытый рот за счет макроглоссии, маленький западающий подбородок, своеобразная походка с неловкими движениями, косноязычие;
• — отставание в психомоторном развитии на первом году жизни;
• — слабоумие;
• — пороки развития сердечно-сосудистой системы (ДМЖП, ОАП);
• — пороки развития желудочно-кишечного тракта (атрезия пищевода);
• — склонность к инфекциям и злокачественным заболеваниям (лейкемия);
• — гипотрофия мышц, увеличение объема движений в суставах, поперечная ладонная складка;
• — пигментные пятна по краю радужки — пятна Брушфильда, косоглазие;
• — невысокий рост, гипотиреоз;
• — аномалии скелета: деформация грудины, укорочение и расширение кистей и стоп, клинодактилия и искривление мизинца, гипоплазия средней его фаланги, сандалевидная щель, может быть единственная складка на 5 пальце, готическое небо, мелкие зубы;
• — крипторхизм, гипоплазия полового члена.
^ (синдром трисомии 18 пары) (Q91.3)
• Кариотип 47 ХХ или ХУ, 18+. Соотношение полов — МI: Ж3. Частота — 1: 8 000 н/д. Трисомию по группе Е впервые описал J. Edwards (1960). Среди новорожденных синдром встречается с частотой около 1:7000, девочки болеют в 3 раза чаще, чем мальчики. И. В. Лурэ, Г. И. Лазюк высказывают мнение о стабилизирующем действие Х-хромосомы при аберрации 18 пары, тогда как зиготы с трисомией 18, имеющие мужской генотип, элиминируются. Возможно также чаще оплодотворения яйцеклетки с лишней хромосомой 18 сперматозоидом, имеет Х-хромосому.Во время цитогенетического обследования в 80% случаев выявляют трисомия 18, а у 10% больных — мозаицизм. Описаны случаи транслокационного варианта, двойной анеуплоидии типа 48, XXV +18 с участием трисомного за хромосомы 18 клона.
Фенотипические проявления синдрома Эдвардса достаточно характерны.
Клиника:
• Отмечаются различные аномалии опорно-двигательного аппарата: грудная клетка расширена, грудина укорочена, таз узкий, конечности деформированы, ограниченная подвижность в тазобедренных суставах, встречается описание вывихов бедра. Кисти и пальцы короткие, дистально расположен и гипоплазирован 1 палец кисти, сглаженный тенар. Пальцы сжаты в кулак за типом «флексорной аномалии» «: II и V пальцы расположены этажа и прикрывают прижаты к ладони III и IV пальцы Первый палец стопы короткий и широкий, синдактилия II и III пальцев. Типичная для трисомии 18 форма стопы в виде» качели «. Характерная общая мышечная гипотония. У мальчиков часто встречается крипторхизм (неопущение яичка в мошонку), гипосподия (аномалия анатомического строения пениса), гипертрофия клитора у девочек.
• Интеллектуальный дефект соответствует олигофрении в стадии идиотии или глубокой имбецильности, и только в редких случаях мозаичного варианта хромосомы 18 умственная отсталость слабее обнаружена. Часто у таких больных развивается судорожный синдром.
• — долихоцефалия, низко посаженные деформированные уши,переносицы вдавленные, но спинка носа тонкая (выступает), ушные раковины расположены очень низко, часто отсутствуют мочка и козелок. выступающий затылок, высокое небо, характерная микроретрогнатия (маленькая и смещенная назад челюсть) короткие глазные щели,рот маленький, треугольной формы с короткой верхней губой, нёбо высокое, иногда с щелью, шея короткая, часто с крыловидной складкой, незаращение губы и неба, микростомия;
• — гипоплазия скелетной мускулатуры и подкожной жировой ткани;
• Погибают в возрасте до 3-5 месяцев, в редких случаях доживают до 5 лет. Синдром Эдвардса (синдром трисомии 18) — второе по частоте после болезни Дауна хромосомное заболевание, характеризуется комплексом множественных пороков развития и трисомией 18 хромосомы
• Интеллектуальный дефект соответствует олигофрении в стадии идиотии или глубокой имбецильности, и только в редких случаях мозаичного варианта хромосомы 18 умственная отсталость слабее обнаружена. Часто у таких больных развивается судорожный синдром.
• Дерматоглифическая картина при синдроме Эдвардса имеет несколько отличительных признаков: большая частота дуг на подушечках пальцев рук (примерно в 10 раз выше, чем в популяции), часто отсутствует дистальная сгибательная складка на пальцах, у трети больных выявляется поперечная ладонная борозда, количество гребешков увеличено, осевой трирадиус обычно расположен дистально.
• На аутопсии при синдроме Эдвардса находят большое количество пороков развития почти всех органов и систем. С разной частотой встречаются аномалии ЦНС: недоразвитость мозолистого тела, мозжечка, атрофия мозговых извилин.
• Почти 95% пациентов с синдромом Эдвардса имеют пороки сердца и крупных сосудов, чаще встречающийся дефект межжелудочковой перегородки и незаращение артериального протока. Около половины всех случаев трисомии 18 сопровождаются врожденными аномалиями органов пищеварения: нарушения размещения кишечника (дивертикул Меккеля), резкое сужение пищевода или анального отверстия. С такой же частотой встречаются пороки развития мочеполовой системы — сегментированная или подковообразная почка, удвоение мочеточников, недоразвитость яичников.
^ (синдром трисомии 13 пары) (Q91.7)
• Кариотип 47 ХХ или ХУ, 13+. Среди больных преобладают девочки.
• Частота 1:10 000 н/д.
• Дети рождаются обычно в срок, но с истинной пренатальной гипоплазией. Наблюдается высокая младенческая смертность (до 90% детей). Часть погибает внутриутробно.
Клиника:
• — микроцефалия;
• — микрофтальм, анофтальмия;
• — одно или двустороннее незаращение верхней губы и неба;
• — полидактилия, выпуклые ногти, поперечная ладонная складка, повышенная гибкость суставов;
• — множественные пороки развития нервной системы и внутренних органов — аплазия мозолистого тела, гипоплазия мозжечка, врожденные пороки сердца (дефект межжелудочковой перегородки, дефект меж предсердной перегородки), аномалии почек (кисты, удвоение ЧЛК, гидронефроз, удвоение мочеточника), пороки развития органов пищеварения (незавершенный поворот кишечника, дивертикул Меккеля);
• — ушные раковины неправильной формы, низко расположены;
• — крипторхизм, гипоплазия наружных половых органов, гипоспадия у мальчиков, удвоение матки и влагалища, двурогая матка у девочек;
• — апноэ;
• судорожный синдром.
Список литературы:
МостюковаЕ.М., Московкина А.Г. Основы генетики :клиннико-генетические основы коррекционной педагогикии специальной психологии.-М.;Владос,2001-192с
А.Ю.Асанов Н.С.Демикова .С.А.Морозов Основы генетики и наследственные нарушения развития у детей :Учеб.пособие для студ.высш. пед.учебных заведений ;-М.;Академия,2003-224с
https://www.4medic.ru/page-id-895.html
(Синдром Дауна:клинодактилия)


(Синдром Дауна -типичность)
Скачать файл (273 kb.)
Источник